fepdualtop
This option specifies the implementation details for single- and dual-topology free-energy perturbation (FEP).
singletopology
Enables single-topology FEP; dual-topology is the default.
block1
Defines the group of atoms that vanish (decouple) during the FEP simulation.
| Value | Atom range |
| Default | None |
block2
Defines the group of atoms that appear (couple) during the FEP simulation.
| Value | Atom range |
| Default | None |
fep_scheme
| Value | pi: parameter interpolation. |
ei: energy interpolation. |
|
| Default | pi |
Defines the strategy for scaling reciprocal-space electrostatic interactions when PME is used. The conventional approach employs energy interpolation: two PME evaluations are performed per time step, and the long-range electrostatic energy of the perturbed system is obtained by combining the two results as follows:
$$U(Col-LR) = (1-\lambda_{col}) U(A-LR) + \lambda_{col} U(B-LR)$$
“LR” stands for long-range electrostatic interactions, while “A”, “B”, and “C” refer to atoms in the vanishing part, appearing part, and environment, respectively.
Alternatively, a more efficient parameter-interpolation scheme can be used to treat long-range electrostatic interactions. In this approach, the force-field parameters—specifically, the charges of the vanishing and appearing atoms—are scaled and evaluated in a single PME calculation:
$$U(Col-LR) = U(LR : q_{C}, \sqrt{\lambda_{col}^A} q_{A}, \sqrt{\lambda_{col}^B} q_{B})$$
neglect_steps
Defines the number of molecular dynamics steps used for system equilibration per lambda window.
Data generated during equilibration will not be recorded.
| Value | An integer |
| Default | 0 |
num_lambda_windows
Defines the number of lambda windows used to transform one end state to the other.
A positive integer is required.
| Value | An integer |
| Default | 2 |
start_window
Defines the index of the lambda window from which the simulation begins.
A positive integer is required.
| Value | An integer |
| Default | 1 |
end_window
Defines the index of the lambda window at which the simulation ends.
| Value | An integer |
| Default | Should be the same as num_lambda_windows |
fepout_freq
The frequency (in MD steps) at which energy components are printed for ensemble averaging.
| Value | An integer |
| Default | 1 |
col_coupledstartwindow
| Value | An integer |
| Default | 0 |
Defines the index of the lambda window after which the electrostatic interactions of appearing atoms begin to be considered.
For vanishing atoms, their electrostatic interactions are gradually scaled to zero from the first window to col_coupledstartwindow (see the illustration below).
The default value 0 disables this keyword, meaning that coupling and decoupling of electrostatic interactions occur across all FEP windows.
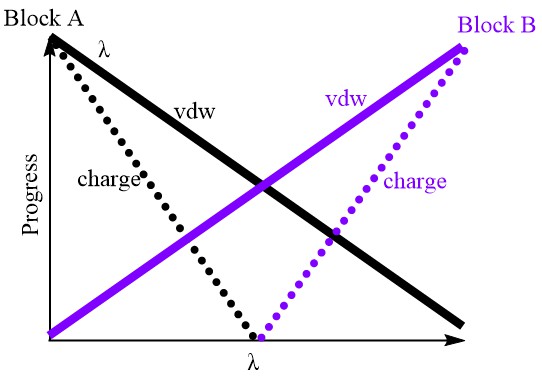
This option is designed to ease the so-called end-point catastrophe problem, which arises from the rapid decay of van der Waals (vdW) interactions compared to electrostatic interactions.
One can imagine that if the vdW interactions between two oppositely charged atoms are scaled close to zero, their remaining significant electrostatic attraction may cause them to overlap, leading to numerical instability.
The logic behind this option is to decouple the electrostatic interactions before the vdW interactions become too weak.
annihilation
Flag used to indicate whether the intra-fragment bonds involving vanishing or appearing atoms are perturbed.
By default, the program assumes no perturbation of intra-fragment bonds.
nointradecouple
Do not decouple the non-bonded interactions between atoms within the vanishing or appearing fragment.
By default, the program does decouple non-bonded interactions within the two end states (i.e., nointradecouple is false).
In some cases, such as determining the relative solvation energy of small and rigid molecules, it is advantageous and saves computational time to set nointradecouple to true.
However, for large and flexible molecules with multiple conformations, it is recommended to leave this option turned off.
lj_lambda_go
Lambda array used to decouple the van der Waals interactions of vanishing atoms with the environment.
If the value of lj_lambda_go is x, the interactions involving block 1 atoms are scaled by (1.0 - x).
The length of the array must match the number of lambda windows.
The same rule applies to lj_lambda_in, col_lambda_go, and col_lambda_in.
lj_lambda_in
Lambda array used to couple the van der Waals interactions of appearing atoms with the environment.
If the value of lj_lambda_in is x, the interactions involving block 2 atoms are scaled by x.
col_lambda_go
Lambda array used to decouple the electrostatic interactions of vanishing atoms with the environment.
col_lambda_in
Lambda array used to couple the electrostatic interactions of appearing atoms with the environment.
The four options — lj_lambda_go, lj_lambda_in, col_lambda_go, and col_lambda_in — provide a flexible way for users to define the lambda transformation pathway.
alpha_soft
| Value | A real number |
| Default | 0. |
Defines the parameter (radius shifting coefficient) used in the modified soft-core van der Waals potential.
The distance between two interacting atoms is shifted from r^2 to r^2 + \alpha * (1 - \lambda) when the interaction is scaled by \lambda.
Do not set an excessively large value (e.g., 20), as it may lead to an unstable or highly distorted interaction potential energy surface.
beta_soft
| Value | A real number |
| Default | 0. |
Defines the parameter (radius shifting coefficient) used in the modified soft-core Coulombic potential.
Similar to alpha_soft for van der Waals interactions.