md
This keyword controls the details of the molecular dynamics (MD) simulation.
Options
type
Define the type of molecular dynamics (MD) simulation.
| Value | NVE: Microcanonical ensemble; constant number of particles, volume, and energy. |
NVT: Canonical ensemble; constant number of particles, volume, and temperature. |
|
fep-NVE: Free energy perturbation calculation for the microcanonical ensemble. |
|
fep-NVT: Free energy perturbation calculation for the canonical ensemble. |
|
| Default | NVE |
dt
The time step in picoseconds (ps). For systems that do not contain hydrogen atoms, dt can be set to 0.001 ps.
| Value | A real number |
| Default | 0.0005 |
num_steps
Specifies the number of MD steps to be run.
The total simulation time is dt × num_steps ps.
| Value | An integer |
| Default | 10 |
output_freq
The number of steps between outputs of MD information.
Trajectory, velocity, and gradient data will be written to disk.
| Value | An integer |
| Default | 1 |
temp
Defines the initial temperature for NVE simulations and the target temperature for NVT simulations.
| Value | A real number |
| Default | 273.15 |
temp_nhc_length
The length of the Nose–Hoover chain used to control the temperature. An integer greater than 2 is usually sufficient.
| Value | An integer |
| Default | 5 |
temp_nhc_tau
The time constant of the Nose–Hoover chain used to control the temperature, in picoseconds (ps).
| Value | A real number |
| Default | 0.5 |
vel_fn
If specified, the initial velocities will be read from the given XYZ file (in Å/ps). Otherwise, velocities will be generated from the Boltzmann distribution.
| Value | A file name |
| Default | None |
For example: initial velocities will be read from mol-vel.xyz.
max_dr
The maximum allowed atomic displacement per step, in Ångström.
If any atom moves more than max_dr in a single step, Qbics will terminate the MD simulation, as such rapid motion may indicate an unstable or crashing system.
| Value | A real number |
| Default | 5 |
plumed_fn
| Value | A file name |
| Default | None |
If specified, Qbics will apply the enhanced sampling algorithms defined in this PLUMED input file.
For the input file syntax, please refer to the PLUMED manual.
Theoretical Background
Molecular Dynamics Simulations
Molecular dynamics (MD) simulations are a computational method used to study the time evolution of a system of interacting particles. In MD simulations, the forces between particles are calculated using a potential energy function, and the equations of motion are solved to obtain the particles' trajectories.
In Qbics, the equations of motion are solved using the velocity-Verlet algorithm, a symplectic integrator that conserves the total energy of the system over long timescales. Forces can be evaluated at various levels of theory, including DFT, MM, NDDO, xTB, DFT/MM, NDDO/MM, and xTB/MM.
During MD simulations, Qbics outputs four different files:
x-md-traj.xyz: The trajectory of the system in XYZ format.x-md-vel.xyz: The velocities of the system in XYZ format.x-md-grad.xyz: The gradients of the system in XYZ format. Note: force is the negative of the gradient.x-md-log.txt: The log file of the MD simulation, containing step number, time, energy components, temperature, and other relevant information.
If plumed_fn is specified, Qbics will also output additional data as defined in the PLUMED input file.
Before running MD simulations, it is strongly recommended to perform a geometry optimization to obtain a stable initial configuration.
Each ensemble has its own conserved quantity.
- For NVE, the total energy is conserved:
$$E = K + V$$
-
Here,
- E is the total energy.
- K is the kinetic energy.
- V is the potential energy.
For example, if you run an NVE MD simulation, you can check the total energy in the file x-md-log.txt:
# Time Total Kinetic Potential Temp Cost
ps Hartree Hartree Hartree Kelvin second
0 0.00000 -34.44404963 0.02280104 -34.46685067 300.00000000 0.257
20 0.01000 -34.44402464 0.01224859 -34.45627323 161.15837759 0.260
40 0.02000 -34.44400898 0.01020550 -34.45421449 134.27682685 0.222
60 0.03000 -34.44401439 0.01628312 -34.46029751 214.24190576 0.220
80 0.04000 -34.44402295 0.02160569 -34.46562864 284.27246073 0.279
100 0.05000 -34.44403491 0.03125266 -34.47528758 411.20057981 0.248
120 0.06000 -34.44402767 0.02061378 -34.46464145 271.22164167 0.228
140 0.07000 -34.44403668 0.02599444 -34.47003112 342.01656058 0.226
160 0.08000 -34.44402275 0.01897475 -34.46299751 249.65643218 0.224- For NVT, the total “virtual” energy is conserved:
$$Q = K + V + \sum_{j=1}^{n_\text{NHC}} \frac{p_{\eta_j}^2}{2Q_j} + 3NkT\eta_1+kT \sum_{j=2}^{n_\text{NHC}}\eta_j$$
-
Here
- nNHC is the Nose–Hoover chain length (
nhc_temp_length). - k is the Boltzmann constant.
- T is the temperature.
- ηj and pηjare the thermostat variables ("position" and "momentum")
- Qjis the thermostat mass (automatically determined by Qbics).
For example, if you run an NVT MD simulation, you can check the ConstQ value in the file x-md-log.txt:
# Time Total Kinetic Potential Temp ConstQ Cost
ps Hartree Hartree Hartree Kelvin Hartree second
0 0.00000 -34.44404963 0.02280104 -34.46685067 300.00000000 -34.44404963 0.000
20 0.01000 -34.44404123 0.01734433 -34.46138557 228.20455596 -34.44404256 0.293
40 0.02000 -34.44401539 0.01535660 -34.45937199 202.05137626 -34.44402161 0.230
60 0.03000 -34.44399789 0.01914301 -34.46314090 251.87027197 -34.44401529 0.213
80 0.04000 -34.44401533 0.03373382 -34.47774915 443.84591789 -34.44404524 0.303
100 0.05000 -34.44399074 0.01808475 -34.46207549 237.94640025 -34.44402711 0.222
120 0.06000 -34.44400165 0.03088255 -34.47488420 406.33083745 -34.44404511 0.218
140 0.07000 -34.44397432 0.01350937 -34.45748370 177.74683071 -34.44402030 0.327
160 0.08000 -34.44397272 0.01886731 -34.46284002 248.24278233 -34.44402353 0.222
180 0.09000 -34.44398418 0.03438731 -34.47837149 452.44411562 -34.44404296 0.225In all cases, if the conserved quantity is not maintained, it indicates that the simulation is unstable. You may need to:
- Reduce the time step;
- Perform a geometry optimization to obtain a stable initial configuration;
- Check whether the potential energy function (quantum chemistry or force field parameters) is appropriate for your system.
However, if plumed_fn is specified, the conserved quantity may not be preserved, as the enhanced sampling algorithm can alter the total energy.
Nose-Hoover Chain algorithm
The Nose–Hoover chain (NHC) algorithm is a method used to control the temperature in MD simulations. It is a generalization of the Nose–Hoover thermostat, consisting of a series of thermostats connected in sequence. The first thermostat controls the temperature of the system, while each subsequent thermostat controls the temperature of the preceding one.
The length of the NHC is controlled by temp_nhc_length, which should be greater than 2.
If temp_nhc_length is set to 1, the system may not produce a correct canonical distribution of velocities.
Input Examples
Example: Dynamic Effects in a Carbocation Reaction
Consider the following reaction:
We want to determine which group is more likely to migrate. To investigate this, we will perform a 200 fs NVT MD simulation starting from the optimized structure of the carbocation. The simulation will be repeated 20 times using a shell script run.sh. Here, we use xTB due to its computational efficiency, though DFT can also be used if sufficient time and resources are available.
The input files are shown below:
mol
C -0.90849 0.15356 -0.48118
C -0.18840 -0.79035 0.24223
C -0.79661 -1.92884 0.73482
C -2.15434 -2.10431 0.51590
C -2.88201 -1.15101 -0.17642
C -2.26735 -0.01407 -0.67971
H -0.22972 -2.66722 1.27887
H -2.64692 -2.98493 0.89733
H -3.93966 -1.29437 -0.33076
H -2.84788 0.72259 -1.21038
C 1.26411 -0.39651 0.38023
C -0.02903 1.30229 -0.93562
C 1.67317 -0.22550 1.85240
H 1.83624 -1.21169 2.27913
H 2.60525 0.32605 1.95669
H 0.88426 0.23363 2.45602
C -0.71228 2.64784 -0.65267
H -0.02047 3.48874 -0.70508
H -1.46470 2.82054 -1.41711
H -1.26222 2.63862 0.29289
C 1.31204 0.95650 -0.38570
H 2.18545 1.61818 -0.54700
C 2.13967 -1.43173 -0.30477
H 1.61435 -2.09539 -0.97595
C 3.43556 -1.57292 -0.10516
H 3.99105 -2.34108 -0.61298
H 4.00084 -0.94936 0.56664
C 0.38001 1.16971 -2.43593
C 1.13470 2.39986 -2.94354
C -0.80675 0.90408 -3.35872
H 1.04783 0.30446 -2.50076
H 1.96481 2.68234 -2.30028
H 1.54266 2.18219 -3.92659
H 0.47031 3.25225 -3.05019
H -1.34333 0.00481 -3.07250
H -1.49736 1.74331 -3.36799
H -0.44263 0.76667 -4.37283
end
basis
def2-svp
end
pseudopotential
def2-ecp
end
scf
charge +1
spin2p1 1
end
xtb
chrg +1
end
md
type nvt
dt 0.001 # 0.001 ps = 1 fs
num_steps 200 # 0.001*200 = 0.2 ps. In real simultion, you may need > 1 ps.
temp 300
temp_nhc_length 5
temp_nhc_tau 0.5
output_freq 10 # 0.001*10 = 10 fs
end
task
md xtb
# md m06 # If you want to use DFT.
end
By bash run.sh, you can run 20 MD simulations. The output files will be md-1-1-md-traj.xyz, etc. We observe that both the isopropyl and vinyl group can migrate. For example:
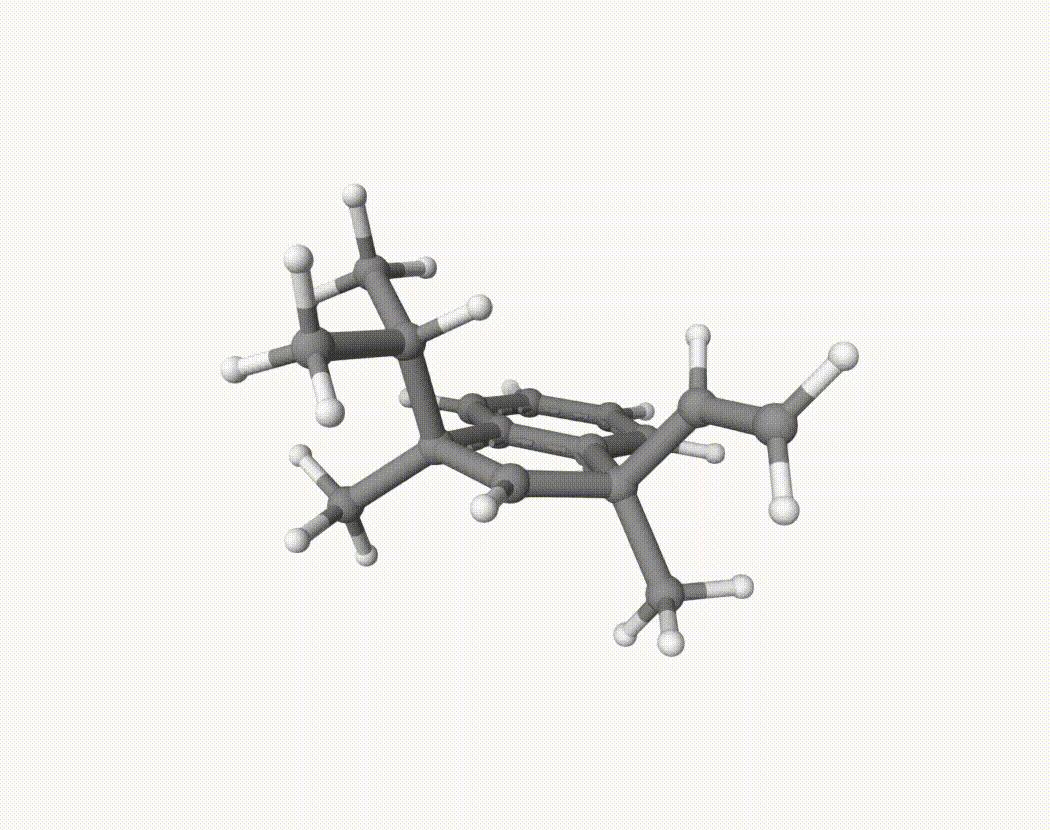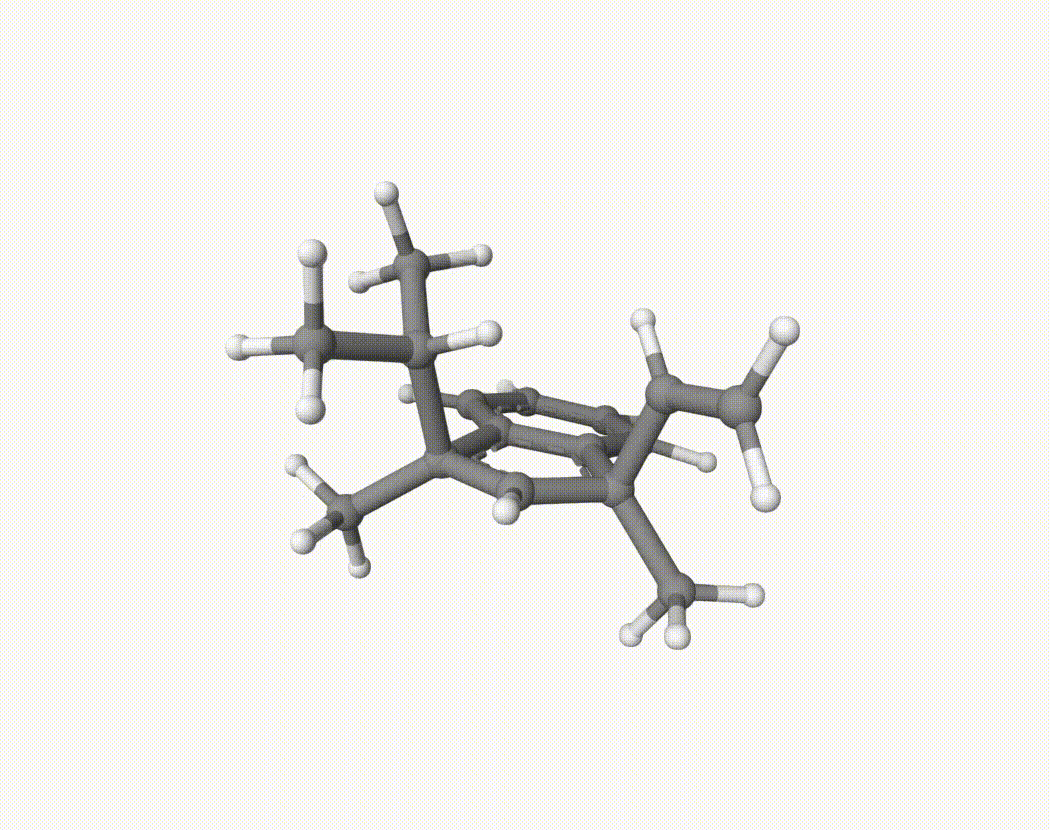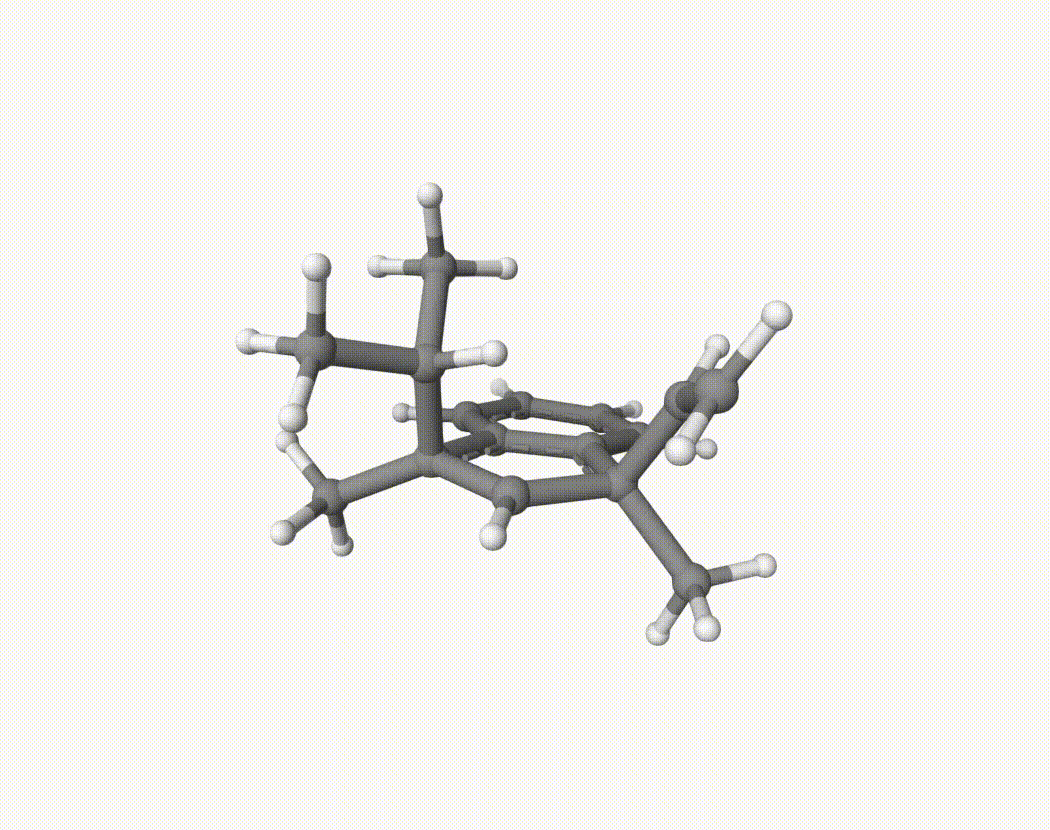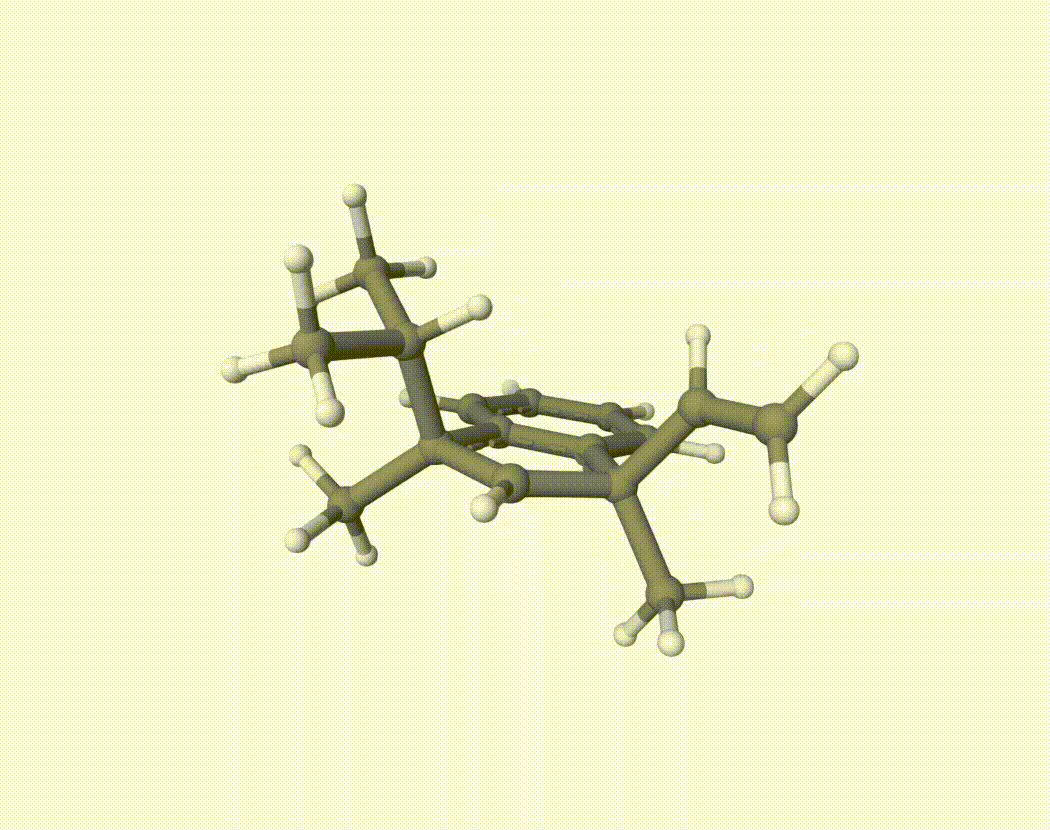
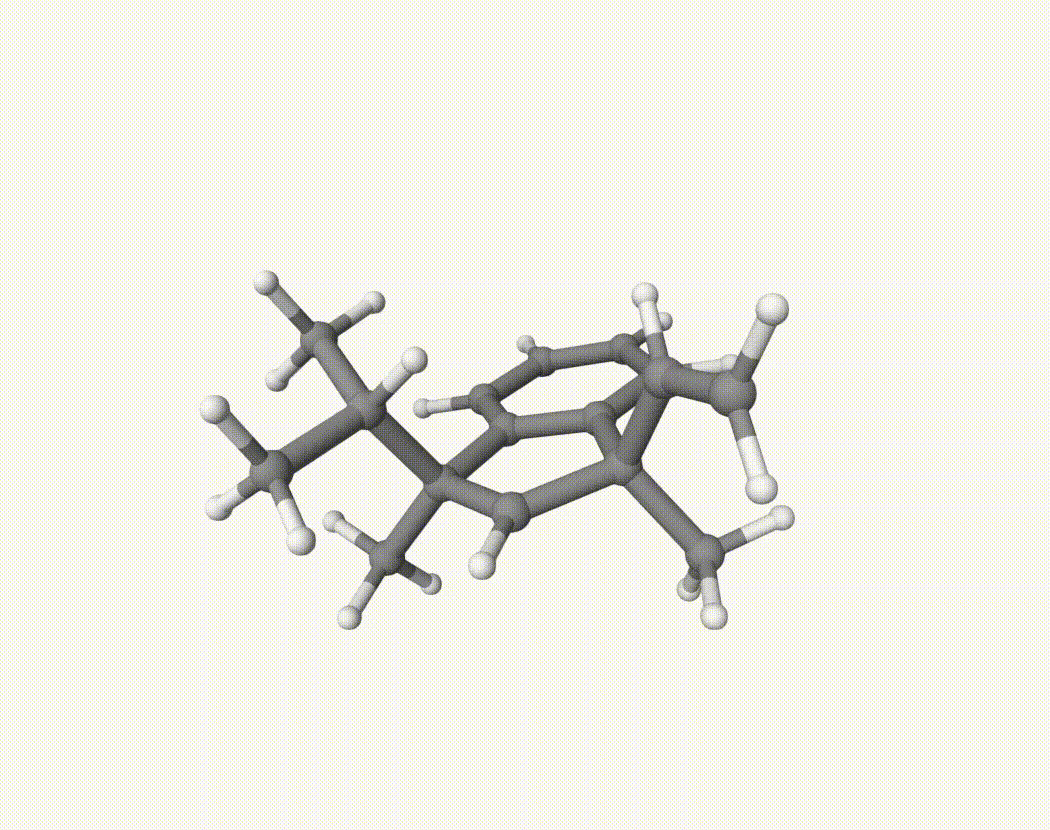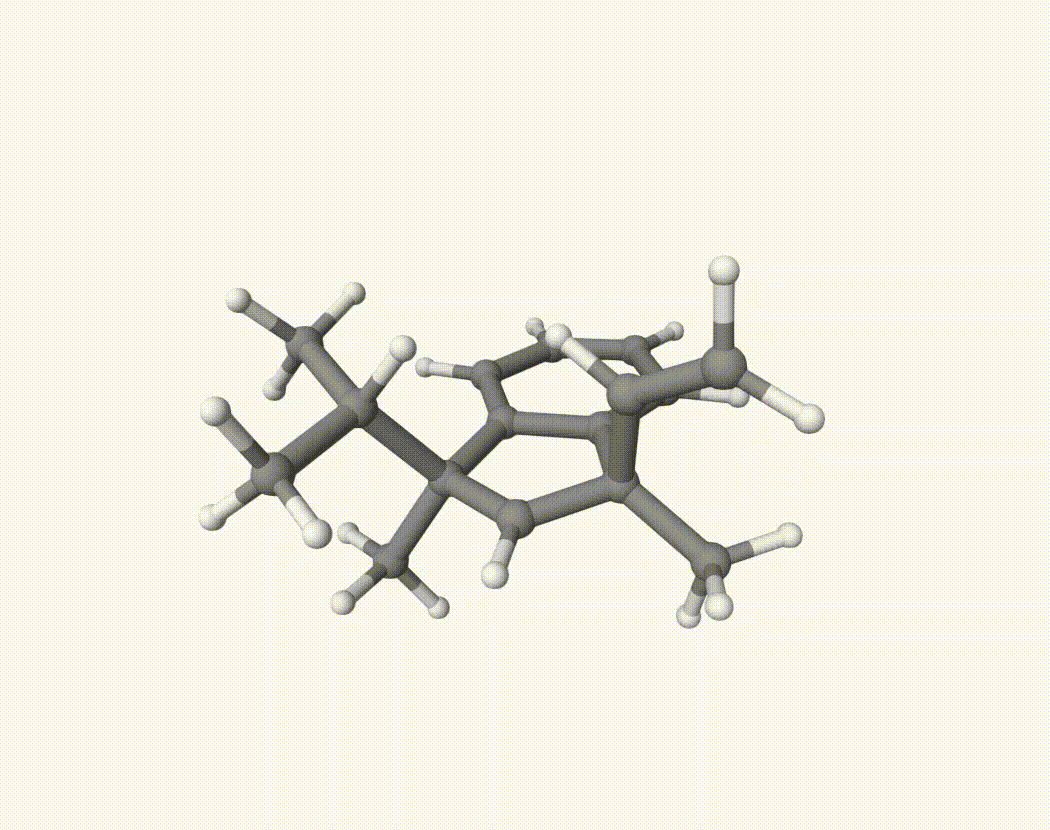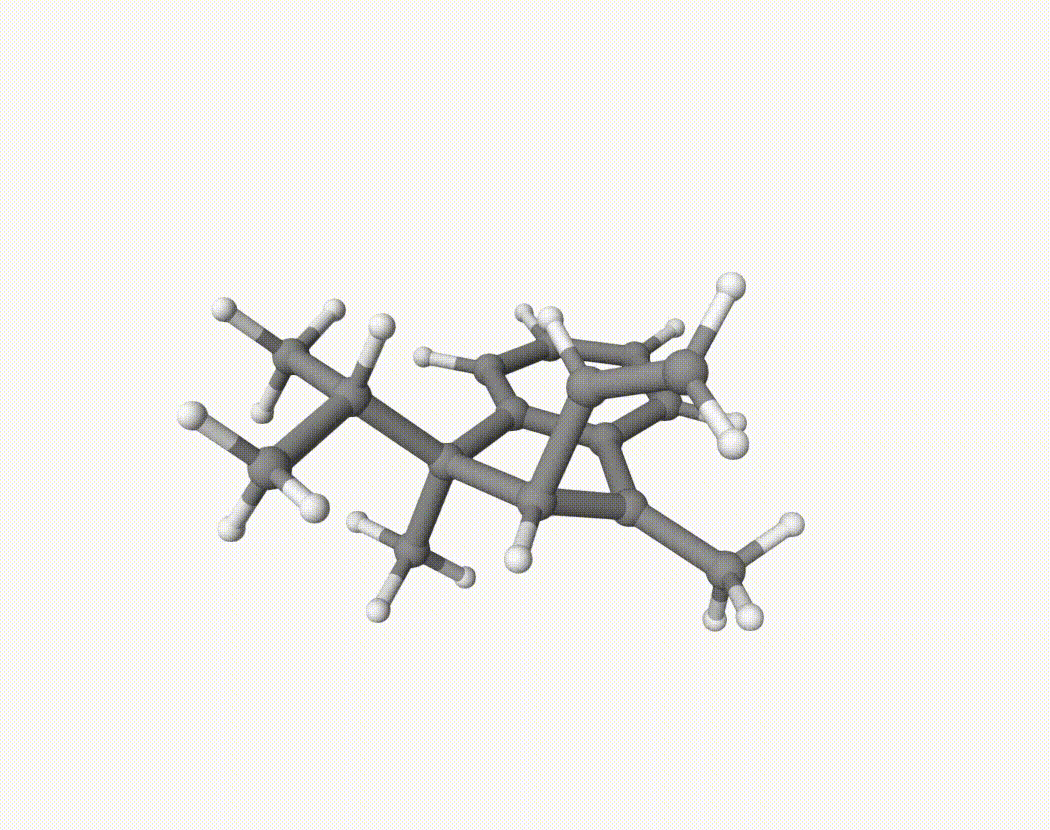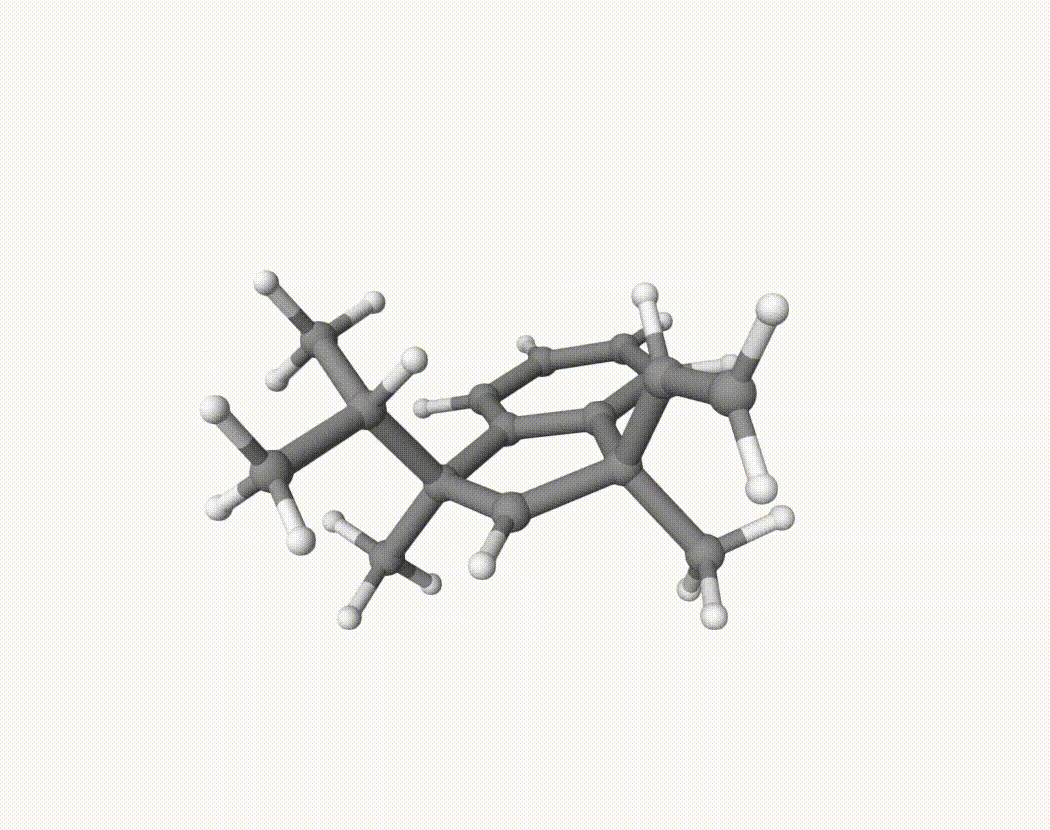
For this test, we calculate the migration ratio of the isopropyl group to the vinyl group.
Based on 20 trajectories, the result is:
| Isopropyl group migration | 7 |
| Vinyl group migration | 7 |
| Non-reactive | 2 |
| Other reactions | 4 |
Of course, if the simulation time is extended, a non-reactive trajectory may become reactive.
Nevertheless, the NVT MD results suggest that the migration ratio is approximately 1:1.
Why do we perform dynamics? Due to the nonstatistical nature of this reaction, the migration ratio cannot be accurately predicted by static calculations.
The results from dynamics are more reliable. For more details, please refer to: J. Am. Chem. Soc. 2021, 143, 1088