charmm
This keyword specifies the implementation details of the CHARMM force field.
Options
parameters
| Value | One or more file names |
| Default | None |
Provide the filenames of the CHARMM force field parameter files (paths may be included).
There is no default file, so at least one filename must be specified.
Multiple files can be listed if needed:
Note: If the same term—for example, a bond like CT-CT—appears in multiple parameter files, the definition in the later file will overwrite those in the earlier ones.
topology
Provide the filename of the molecule's topology file in PSF format (path may be included).
| Value | A file name |
| Default | None |
scaling14
Define the scaling factor for electrostatic interactions of the 1–4 term. In most cases, this value is set to 1.0.
| Value | A real number |
| Default | 1.0 |
rcutoff
Define the cutoff distance for non-bonded interactions, in angstroms.
When use_PBC is enabled, rcutoff must be smaller than half the size of the periodic box to avoid self-interactions.
If rswitch is greater than rcutoff, the switching function will not be applied, and no cutoff will be used for long-range forces.
| Value | A real number |
| Default | 15.0 |
rswitch
Define the distance (in angstroms) at which the switching function is activated.
If rswitch is greater than rcutoff, the switching function will be disabled, and no cutoff will be applied to long-range forces.
| Value | A real number |
| Default | 10.0 |
use_PBC
Enable periodic boundary conditions (PBC) for the system.
If this option is not specified, the system will be treated as non-periodic (i.e., in the gas phase).
Lbox
When use_PBC is enabled, this option specifies the lattice lengths along the X, Y, and Z directions in angstroms.
| Value | Three real numbers |
| Default | 0. 0. 0. |
For example, the following creates a simulation box of 86 Å × 86 Å × 86 Å.
electrostatic
-
Specify the scheme used to calculate electrostatic interactions.
- For gas-phase systems,
Cutoffis the only available method. - For MM calculations in periodic systems, both
CutoffandPMEare supported, withPMErecommended. - For QM/MM calculations in either gas-phase or periodic systems, only
Cutoffis currently supported.
| Value | Cutoff Cutoff scheme for gas phase or periodic systems. |
PME Particle mesh Ewald (PME) method. Only available for periodic systems. |
|
| Default | Cutoff |
PMEk
Specify the number of grid points for PME long-range force calculations.
It is recommended to choose integers close to the lattice lengths that are divisible only by 2, 3, and/or 5.
For example, if the lattice box size (Lbox) is 109 × 109 × 77, then PMEk can be set to 108 108 80.
| Value | Three integers |
| Default | 64 64 64 |
Theoretical Background
CHARMM Force Field
-
To perform a CHARMM force field calculation, in addition to system coordinates,
one must provide the system topology (via the
- Bonded interactions include:
- Bond stretching
- Angle bending
- Dihedral angle rotation
- Improper torsion
- Nonbonded interactions include:
- Lennard-Jones interactions
- Electrostatic interactions
topology keyword) and force field parameter files
(via the parameters keyword). The CHARMM force field consists of bonded and nonbonded interactions.
For nonbonded interactions in non-periodic systems, all pairwise interactions can be computed by setting rswitch larger than rcutoff.
However, computing all pairwise interactions can be computationally expensive.
In such cases—both for non-periodic and periodic systems—a cutoff scheme can be used to truncate long-range interactions.
In the cutoff scheme (electrostatic cutoff), interactions are truncated at a specified distance rcutoff.
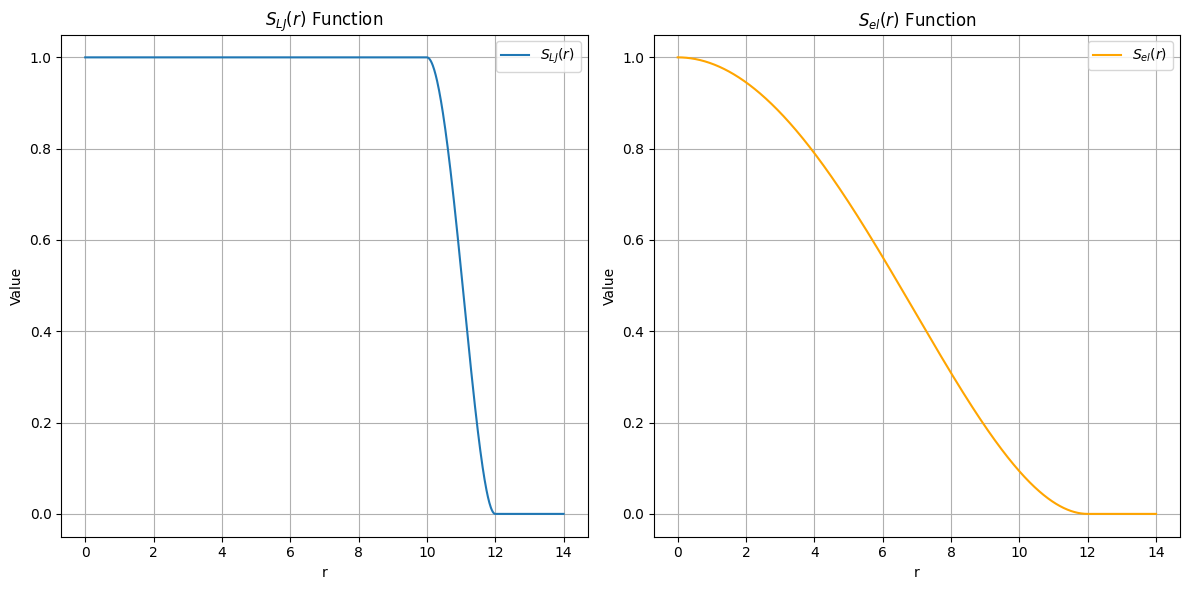
A switching function is then applied to smoothly reduce the interaction strength starting from a distance rswitch. Note that the switching functions differ between Lennard-Jones and electrostatic interactions.
Their mathematical expressions are shown below:
$$\begin{aligned}
S_{\text{LJ}}(r) =
\begin{cases}
1 & \text{if } r \le r_\text{s} \\
\frac{\left(r_\text{c}^{2}-r^{2}\right)^2\left(r_\text{c}^{2}-r^{2}-3\left(r_\text{s}^{2}-r^{2}\right)\right)}{\left(r_\text{c}^{2}-r_\text{s}^{2}\right)^3} & \text{if } r_\text{s} \le r \le r_\text{c} \\
0 & \text{if } r \ge r_\text{c} \\
\end{cases}
\end{aligned}$$
$$\begin{aligned}
S_{\text{el}}(r) = \begin{cases}
\left(1-\frac{r^2}{r_\text{c}^{2}}\right)^2 & \text{if } r \le r_\text{c}\\
0 & \text{if } r \le r_\text{c} \\
\end{cases}
\end{aligned}$$
Plots of these functions are shown below.
For periodic systems, the Particle Mesh Ewald (PME) method (specified as electrostatic pme) can be used to calculate long-range electrostatic interactions.
Additionally, rcutoff should be smaller than half the size of the periodic box to avoid self-interactions.
PSF File Format
A Protein Structure File (PSF) is a file format used to describe the topological information of biological macromolecules. PSF files primarily store data related to atoms, bonds, angles, dihedral angles, and improper force terms. However, they do not contain atomic coordinates.
The PSF file consists of the following sections relevant to Qbics:
- Atoms: Lists each atom’s ID, residue information, atom type, charge, mass, etc.
- Bonds: Defines covalent bonds between atoms. Each line contains the IDs of atom pairs.
- Angles: Records bond angles formed by three atoms. Each line contains three atomic IDs.
- Dihedrals: Defines dihedral (torsion) angles involving four atoms. Each line includes four atomic IDs.
- Impropers: Represents mechanical constraints used to preserve molecular planarity (e.g., the coplanar structure of a benzene ring). Each line contains four atomic IDs.
6 !NTITLE
REMARKS original generated structure x-plor psf file
REMARKS 2 patches were applied to the molecule.
REMARKS topology top_all27_prot_lipid.inp
REMARKS segment U { first NTER; last CTER; auto angles dihedrals }
REMARKS defaultpatch NTER U:1
REMARKS defaultpatch CTER U:76
1231 !NATOM
1 U 1 MET N NH3 -0.300000 14.0070 0
2 U 1 MET HT1 HC 0.330000 1.0080 0
3 U 1 MET HT2 HC 0.330000 1.0080 0
4 U 1 MET HT3 HC 0.330000 1.0080 0
5 U 1 MET CA CT1 0.210000 12.0110 0
6 U 1 MET HA HB 0.100000 1.0080 0
7 U 1 MET CB CT2 -0.180000 12.0110 0Key Sections of a PSF File
- Title Section: Contains annotation information of the PSF file, such as generation method, topology file path, and so on.
- Atoms Section:
- Atom ID: A unique identifier (e.g., 1, 2).
- Segment Name: For example,
Ufor a protein chain. - Residue ID: The ID of the residue to which the atom belongs (e.g., 1 for the first residue).
- Residue Name: For example,
METfor methionine. - Atom Name: Atom label, such as
N,HT1,HT2,CA, etc. - Atom Type: The type used in force field calculations (e.g.,
NH3,HC,CT1). - Charge: Partial charge of the atom (e.g., -0.300000).
- Mass: Atomic mass (e.g., 14.0070).
-
Bonds, Angles, Dihedrals, and Impropers Sections:
These follow the Atoms section and contain corresponding structural information.
- Bonds: Lists pairs of atom IDs that are covalently bonded.
- Angles: Lists triplets of atom IDs defining bond angles.
- Dihedrals: Lists quadruplets of atom IDs defining torsional angles.
- Impropers: Lists quadruplets of atom IDs used to enforce planarity or chirality.
7517 !NBOND: bonds
1 2 1 6 1 7 2 3
2 12 3 4 3 8 4 5
4 9 5 6 5 10 6 11
7 13 7 14 12 13 14 15
14 16 17 18 17 19 17 20
17 21 19 22 23 24 23 25
23 26 23 27 25 28 29 30Here, we have bonds such as 1–2, 1–6, 1–7, 2–3, and so on. The format is similar for the other sections.
CHARMM Parameter File Format
CHARMM force field files define the rules and parameters governing interactions between atoms in a molecular system. The file contains several sections relevant to Qbics:
- BONDS: Defines the equilibrium lengths of covalent bonds and their corresponding force constants.
- ANGLES: Provides the equilibrium values of bond angles and their associated force constants.
- DIHEDRALS: Specifies the potential energy function and parameters for torsional angles.
- IMPROPERS: Specifies the potential energy function and parameters for out-of-plane (improper) torsions.
- NONBONDED: Defines the Lennard-Jones potential for nonbonded interactions.
Other sections, such as NBFIX, are not used by Qbics.
Input Examples
Example: Molecular Dynamics Simulation of Ubiquitin in Water
Below is an example Qbics input file for simulating the protein ubiquitin in water. The system includes a total of 24,696 atoms—ubiquitin solvated in a 64 × 64 × 64 Å3 water box.
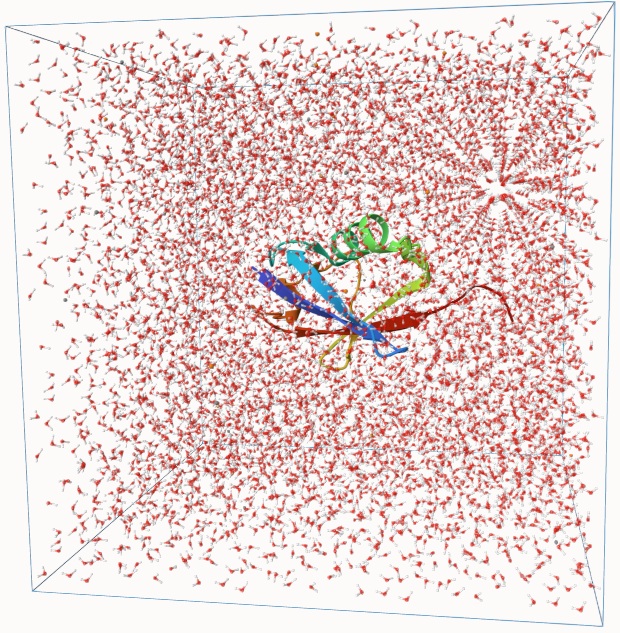
This system was built by randomly placing water molecules around the ubiquitin.
This usually results in a high-energy configuration, and running molecular dynamics directly on it can lead to instability.
Therefore, we first perform energy minimization to optimize the structure:
charmm
# Here:
# toppar_water_ions.str: the parameter file for water and ions.
# par_all36m_prot.prm: the parameter file for proteins.
# Both are obtained from website: https://mackerell.umaryland.edu/charmm_ff.shtml.
parameters toppar_water_ions.str par_all36m_prot.prm
topology sys.psf
scaling14 1.0
rcutoff 12.0
rswitch 10.0
use_PBC # Add this line to turn on PBC.
Lbox 64 64 64 # Define the box size.
electrostatic pme # Use PME for electrostatics.
PMEk 64 64 64
end
opt
max_step 500
end
mol
sys.pdb
end
task
opt charmm
endAfter 500 steps of energy minimization, the system’s total energy decreased from 34,167,803.38 kcal/mol to -82,402.26 kcal/mol.
Using the optimized structure, we can now proceed with an NVT molecular dynamics (MD) simulation:
charmm
# Here:
# toppar_water_ions.str: the parameter file for water and ions.
# par_all36m_prot.prm: the parameter file for proteins.
# Both are obtained from website: https://mackerell.umaryland.edu/charmm_ff.shtml.
parameters toppar_water_ions.str par_all36m_prot.prm
topology sys.psf
scaling14 1.0
rcutoff 12.0
rswitch 10.0
use_PBC # Add this line to turn on PBC.
Lbox 64 64 64 # Define the box size.
electrostatic pme # Use PME for electrostatics.
PMEk 64 64 64
end
md
type nvt
dt 0.001 # 1 fs
num_steps 1000 # 1 ps. Just for demonstration.
output_freq 100
temp 298
end
mol
charmm-1a-opt.xyz # Use the optimized structure from the previous step.
end
task
md charmm
end