opt
This keyword controls the search for geometry minima, transition states, and reaction paths.
Options
type
| Value | Min: Searches for the geometry minimum. |
NEB: Uses the Nudged Elastic Band (NEB) algorithm to search for the reaction path and transition state. |
|
Dimer: Uses the dimer algorithm to locate the transition state, given the reactant and product geometries.
|
|
| Default | Min |
Specify the type of geometry optimization to perform:
-
For
Min, the initial structure must be provided inmol. The optimization process is saved tox-opt-traj.xyz, and the final optimized minimum is written tox-opt.xyz. -
For
NEBandDimer, two structures must be provided inmolandmol2, representing the reactant and product poses, respectively. Typically,NEBis used first to quickly find a reasonable path and transition state. IfNEBfails to converge, the resulting structures can then be used in aDimersearch.Dimeris computationally cheaper thanNEB, but it requires high-quality reactant and product structures. -
For
NEB, the reaction path is saved tox-opt-traj.xyz, and the final transition state is written tox-opt.xyz. -
For
Dimer, the transition state is saved tox-opt.xyz. Note thatx-opt-traj.xyzis not the reaction path—it simply records the optimization steps, similar toMin.
max_step
The maximum number of geometry optimization steps.
| Value | An integer |
| Default | 1000 |
energy_cov
The energy convergence threshold. When the energy change falls below this value, the energy criterion is considered satisfied.
| Value | A real number |
| Default | 1.E-4 |
grad_cov
| Value | A real number |
| Default | 1.E-3 |
The gradient convergence threshold. This value determines four specific convergence criteria:
| Maximum gradient component | grad_cov |
| RMS gradient: | grad_cov * 2/3 |
| Maximum atomic displacement | grad_cov * 4 |
| RMS atomic displacement | grad_cov * 8/3 |
When all four conditions are met, the gradient criterion is considered satisfied.
max_dr
The maximum atomic displacement allowed in a single optimization step.
If the molecule is highly flexible (i.e., the potential energy surface is very flat), or if the structure—particularly a transition state—is close to a stationary point but not yet converged, setting a smaller max_dr value (e.g., 0.1) can be very helpful.
| Value | A real number |
| Default | 0.5 |
num_images
The number of images used in the NEB transition state search.
This number should not be set too small (e.g., 5), as it may lead to an inaccurate reaction path.
| Value | An integer |
| Default | 10 |
neb_k
| Value | A real number |
| Default | 0.01 |
The force constant used in NEB transition state search.
For a given system, the optimal value of neb_k should be determined through trial and error.
fix_atoms
| Value | Atom range |
| Default | None |
Specifies the atoms to be fixed during geometry optimization.
For example: atoms 2, 5, 6, 7, 8, 9, 23, and 26 will remain fixed throughout the optimization.
fix_bond
| Value | 2 integers |
| Default | None |
Specifies the bonds to be fixed during geometry optimization.
For example: bonds (1, 4) and (2, 6) will remain fixed throughout the optimization.
fix_angle
| Value | 3 integers |
| Default | None |
Specifies the angles to be fixed during geometry optimization.
For example: angles (1, 4, 5) and (2, 6, 7) will remain fixed throughout the optimization.
fix_torsion
| Value | 4 integers |
| Default | None |
Specifies the torsions to be fixed during geometry optimization. For example: torsions (1, 4, 5, 9) and (2, 6, 7, 12) will remain fixed throughout the optimization.
Theoretical Background
Minimum
A minimum is defined as a stable isomer on the potential energy surface (PES) of a molecule, where the gradients on all atoms are zero.
The optimization result strongly depends on the initial structure; different starting geometries may lead to different isomers.
Transition State
A transition state is a short-lived atomic configuration that represents a maximum along one direction and a minimum along all others on the potential energy surface. The gradients on all atoms are also zero.
In Qbics, the transition state can be located using the NEB or dimer method. Only the (unoptimized) reactant and product structures are required—no exact Hessian needs to be computed.
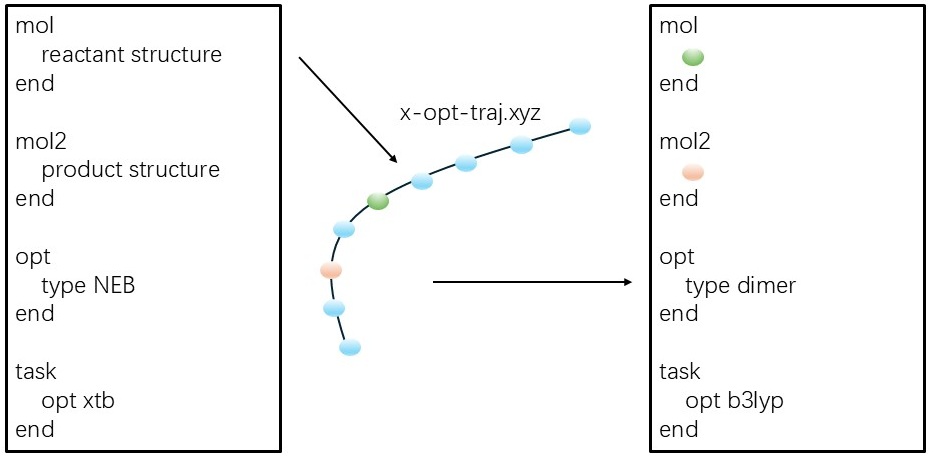
A recommended strategy is:
-
Use a low-cost method, such as xTB, to identify a reasonable reaction path and transition state using the NEB method (
type neb). -
Then, refine the transition state using a standard DFT method with the dimer approach (
type dimer), even if the initial NEB result has not fully converged.
This strategy is illustrated below:
Input Examples
Example: Minimum Structure of Aspirin
Search for the minimum structure of aspirin at the B3LYP/def2-SVP level of theory:
basis
def2-svp
end
scf
charge 0
spin2p1 1
end
mol
O 1.23330 0.55400 0.77920
O -0.69520 -2.71480 -0.75020
O 0.79580 -2.18430 0.86850
O 1.78130 0.81050 -1.48210
C -0.08570 0.60880 0.44030
C -0.79270 -0.55150 0.12440
C -0.72880 1.84640 0.41330
C -2.14260 -0.47410 -0.21840
C -2.07870 1.92380 0.07060
C -2.78550 0.76360 -0.24530
C -0.14090 -1.85360 0.14770
C 2.10940 0.67150 -0.31130
C 3.53050 0.59960 0.16350
H -0.18510 2.75450 0.65930
H -2.72470 -1.36050 -0.45640
H -2.57970 2.88720 0.05060
H -3.83740 0.82380 -0.50900
H 3.72900 1.41840 0.85930
H 4.20450 0.69690 -0.69240
H 3.71050 -0.36590 0.64260
H -0.25550 -3.59160 -0.73370
end
task
opt b3lyp
endExample: Minimum Structure with Constraints
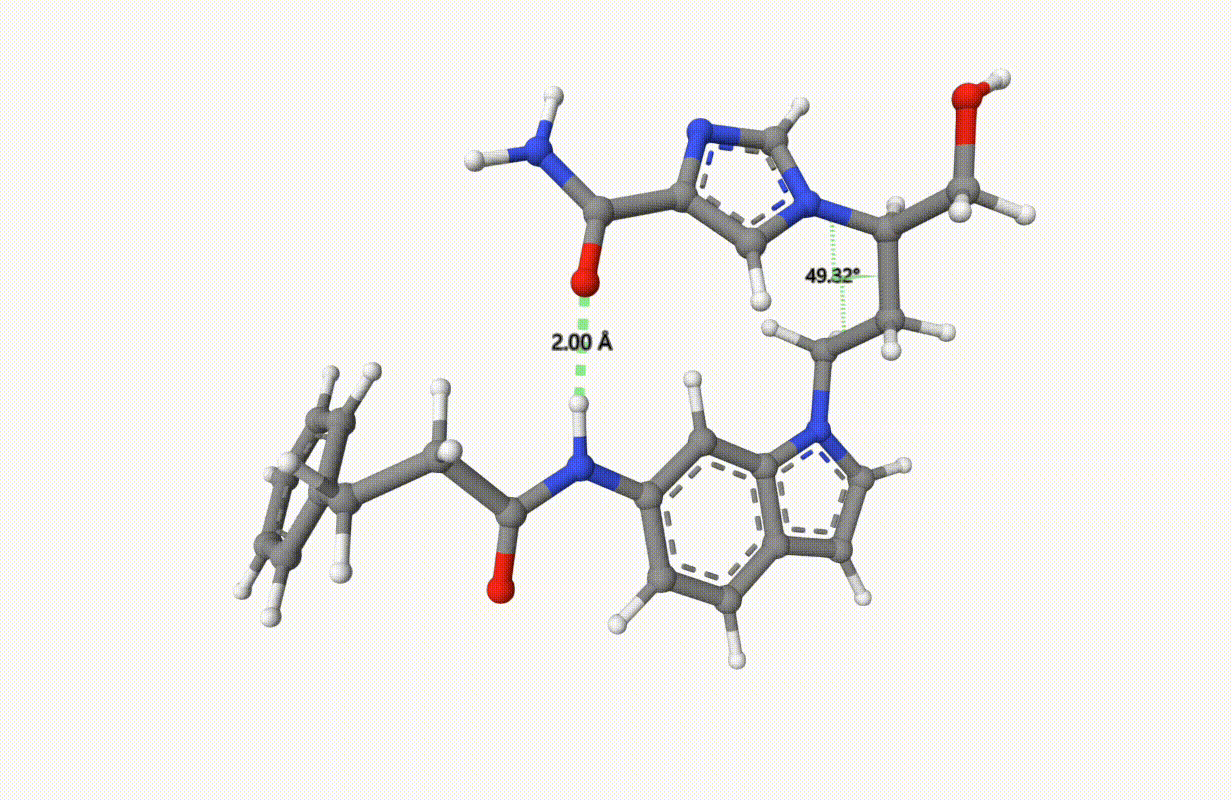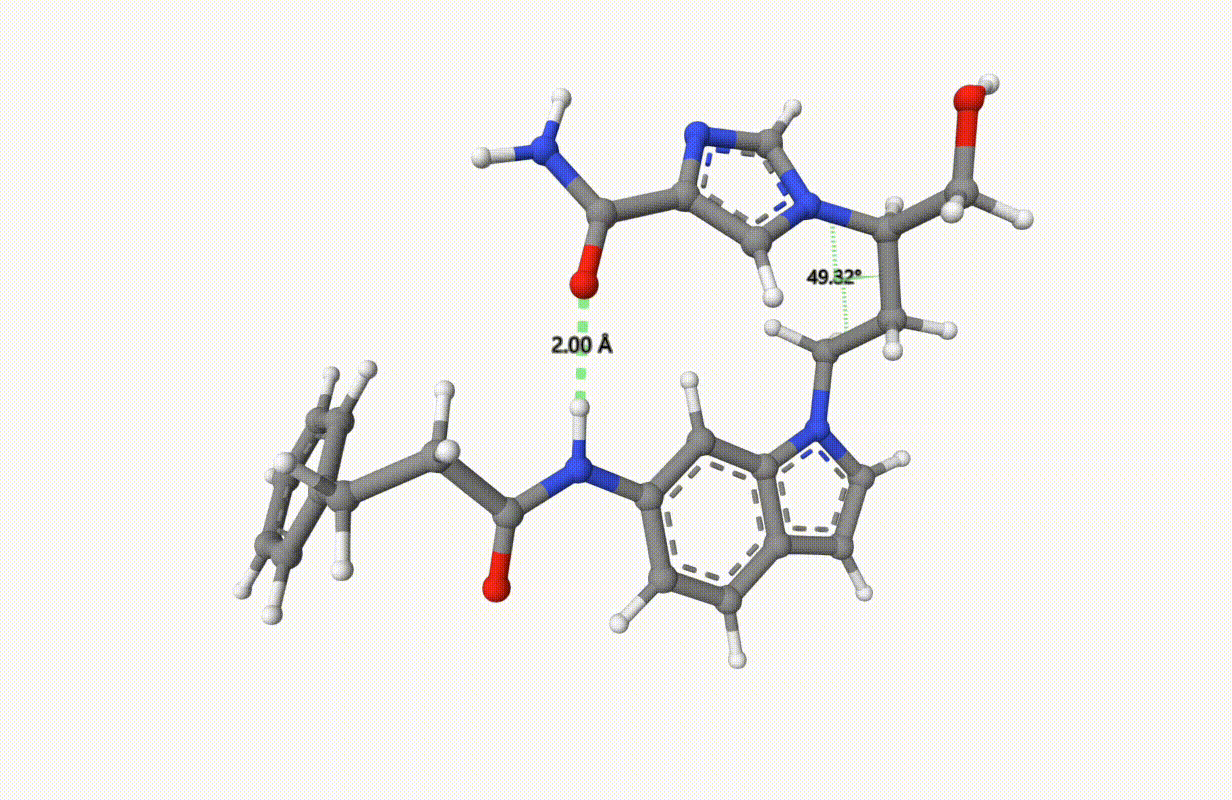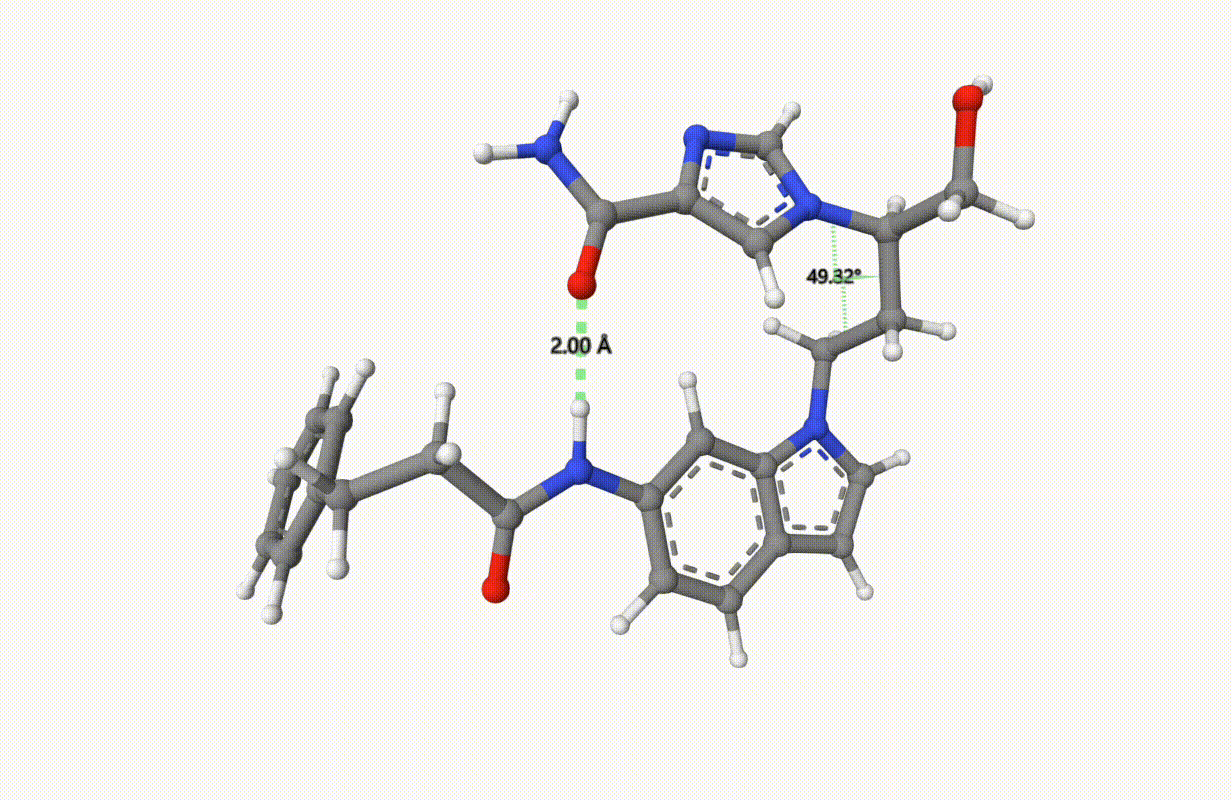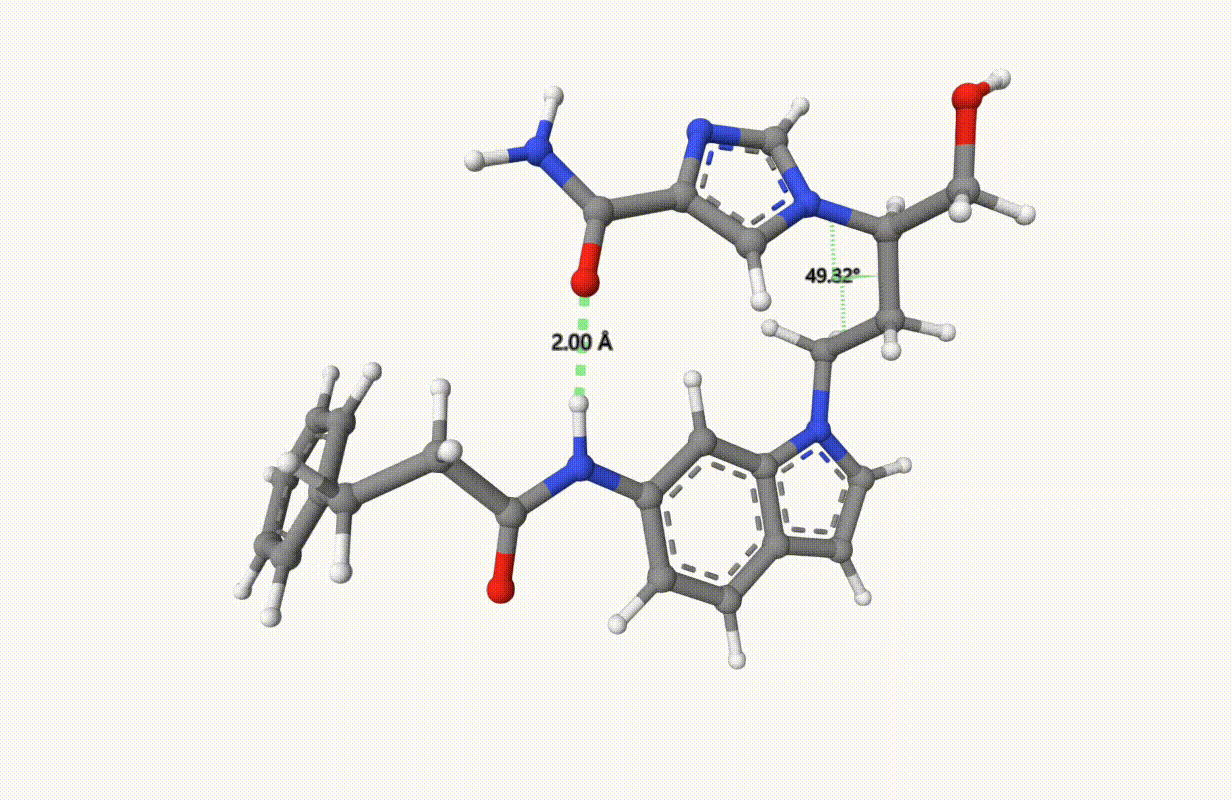
Search for the minimum structure of the molecule "1UML" with one bond and one torsion fixed, using the xTB level of theory:
Check the applied constraints during the optimization:
Example: Transition State of SN2 Reaction
Search for the transition state of an SN2 reaction using the NEB algorithm at the B3LYP/def2-SVP level of theory:
basis # Define basis set.
def2-svp
end
opt
type NEB # Type: Min, NEB, Dimer
num_images 10 # The number of images for NEB calculations.
neb_k 0.01 # The force constant for NEB calculations.
end
scf
charge -1 # The net charge.
spin2p1 1 # 2S+1
end
xtb
chrg -1
end
mol
C -2.25147439 4.89406277 -1.00469981
H -1.89481996 3.88525277 -1.00469981
H -1.89480154 5.39846096 -0.13104831
H -3.32147439 4.89407596 -1.00469981
Cl -1.66479756 5.72372709 -2.44173406
Cl -2.67350651 4.09697871 0.73250622
end
mol2
C -2.36845504 4.69197207 -0.60149770
H -1.76657311 4.00286639 -1.15626927
H -1.80200132 5.57659799 -0.39786281
H -3.23625780 4.94775799 -1.17280492
Cl -1.66479756 5.72372709 -2.44173406
Cl -2.86278952 3.94963672 0.91579319
end
task
opt b3lyp
# opt xtb # You can also try this.
endThe reaction path is provided in ts-1-opt-traj.xyz:
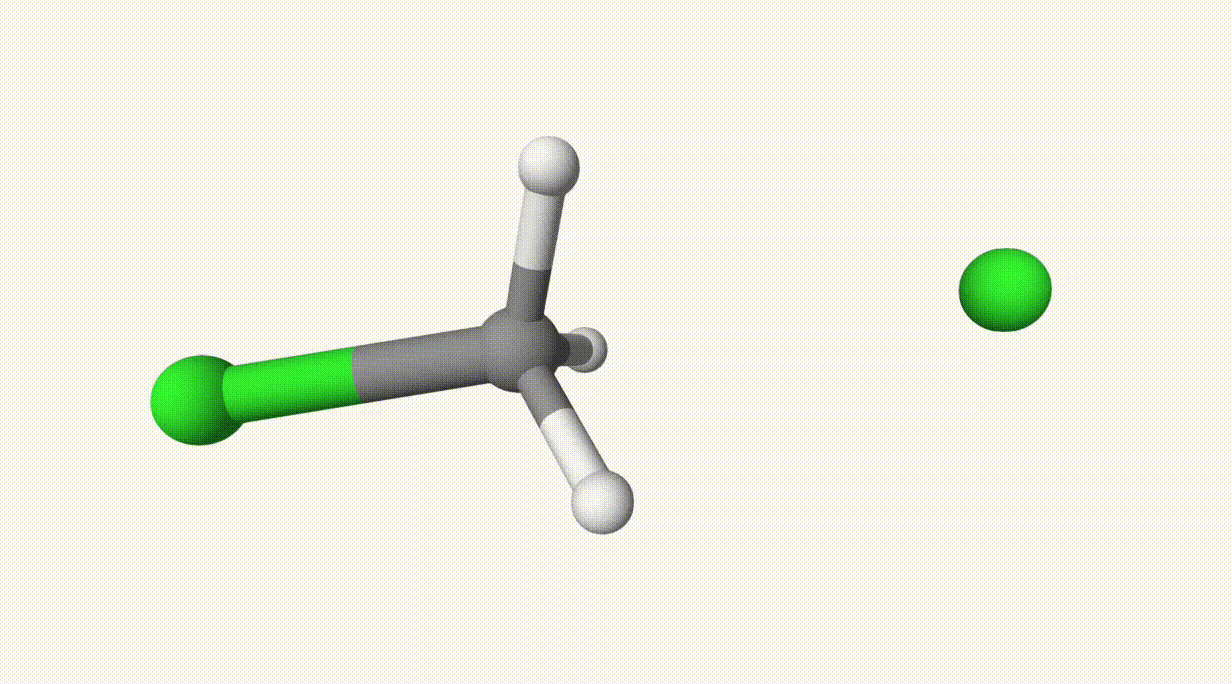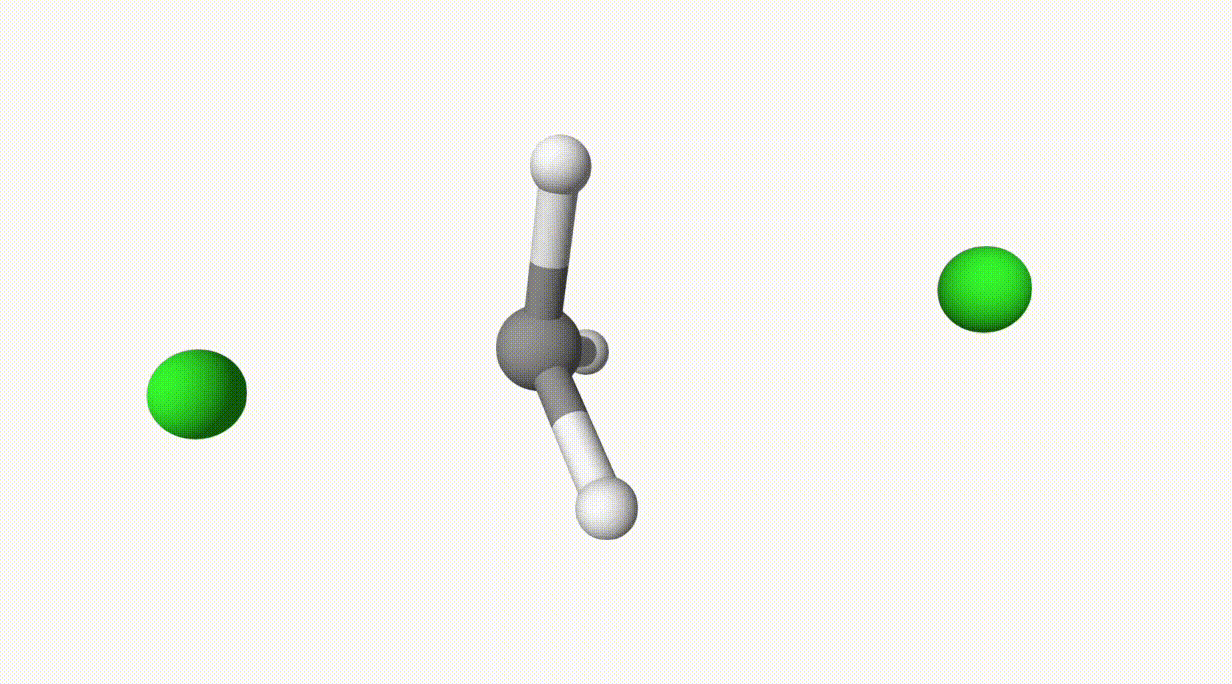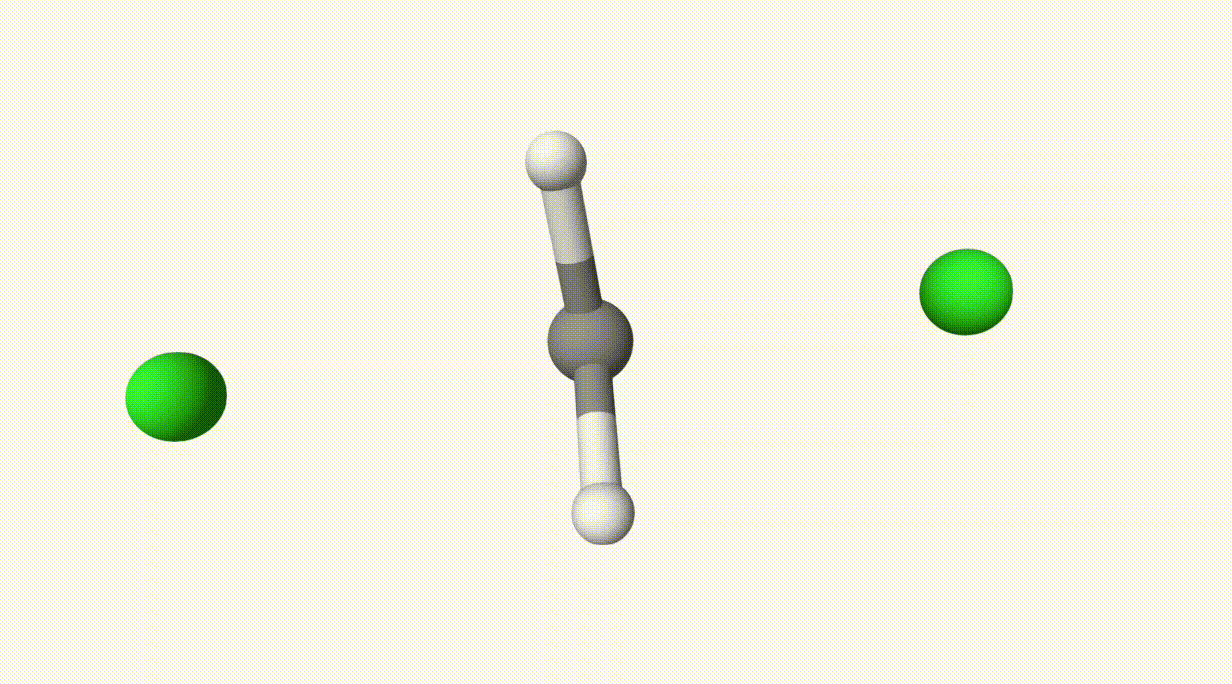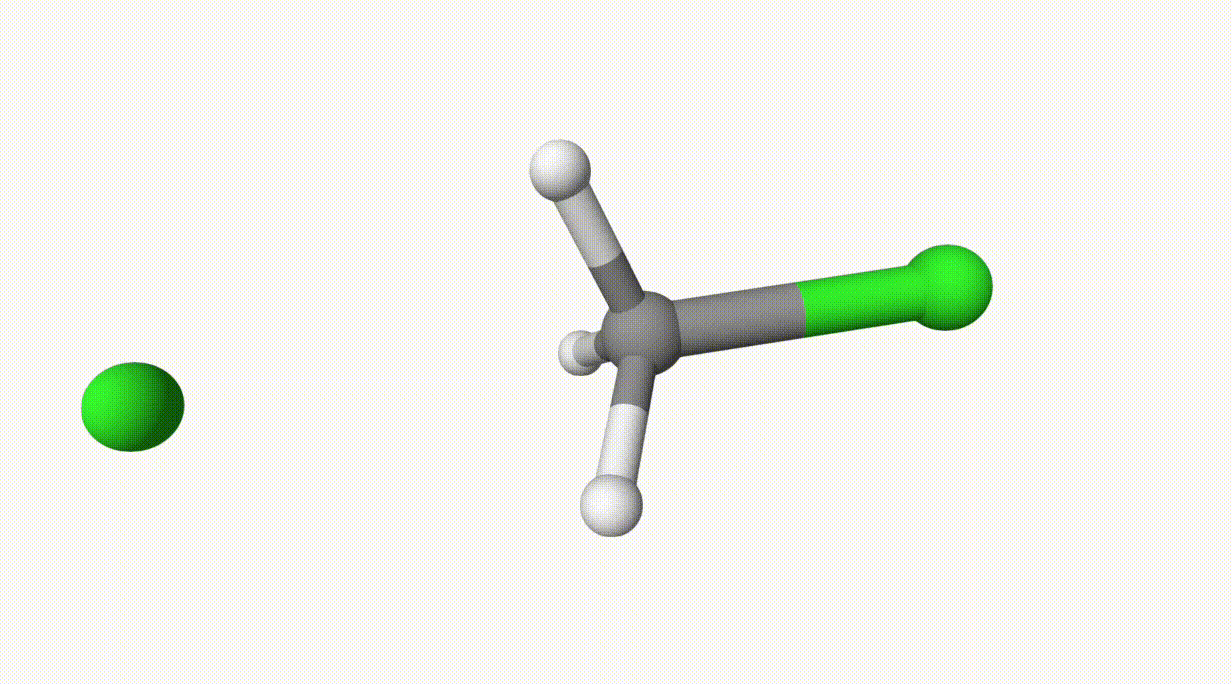
The energies can be found in the output file ts-1.out:
NEB path updated in "ts-1-opt-traj.xyz":
----------------------------------------------------
# Energy Dist Gtang Gperp
----------------------------------------------------
0 -960.06873748 0.13968 0.00000 0.00000
1 -960.06864683 0.11091 0.00029 0.00035
2 -960.06738628 0.07888 0.00032 0.00028
3 -960.06508634 0.07078 0.00008 0.00030
4 -960.06240481 0.09854 -0.00028 0.00036
5 -960.05912811 0.18516 -0.00087 0.00032
6 -960.05781339 0.21042 -0.00025 0.00051
7 -960.06424460 0.26756 -0.00057 0.00032
8 -960.06880165 0.00000 0.00000 0.00000
9 -960.05738746 0.06067 0.00014 0.00026
----------------------------------------------------
Geometry optimization step 34:
Current energy: -960.05738746
Delta Energy: 8.34686E-08; Target: 1.00000E-04; Converged? Yes
Max displacement: 2.30167E-04; Target: 4.00000E-03; Converged? Yes
RMS displacement: 1.07545E-04; Target: 2.66667E-03; Converged? Yes
Max gradient: 5.45965E-04; Target: 1.00000E-03; Converged? Yes
RMS gradient: 2.56874E-04; Target: 6.66667E-04; Converged? Yes
Stationary point has reached.
In the table, structure 0 and 1 correspond to the reactant and product, respectively. Structure 6 represents the transition state, which is also saved in ts-1-opt.xyz.
You can replace b3lyp with xtb to perform the calculation more quickly.
You can also try type dimer, which is computationally cheaper than NEB, but requires high-quality reactant and product structures.
Example: Transion State of Decarboxylation Reaction
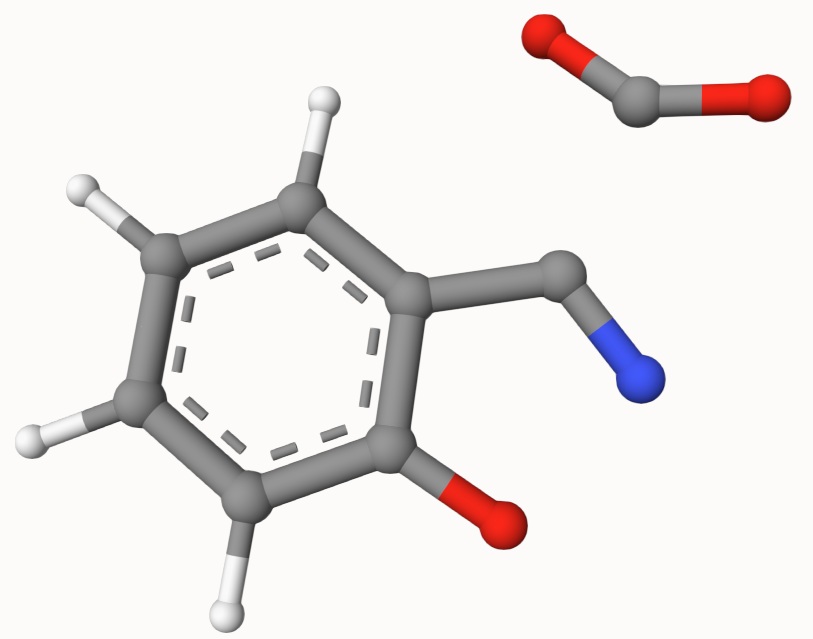
Search for the transition state of the following decarboxylation reaction using the NEB algorithm at the B3LYP/def2-SVP level of theory:
The structures in a1.xyz and a2.xyz are shown below. They are placed arbitrarily without prior optimization.
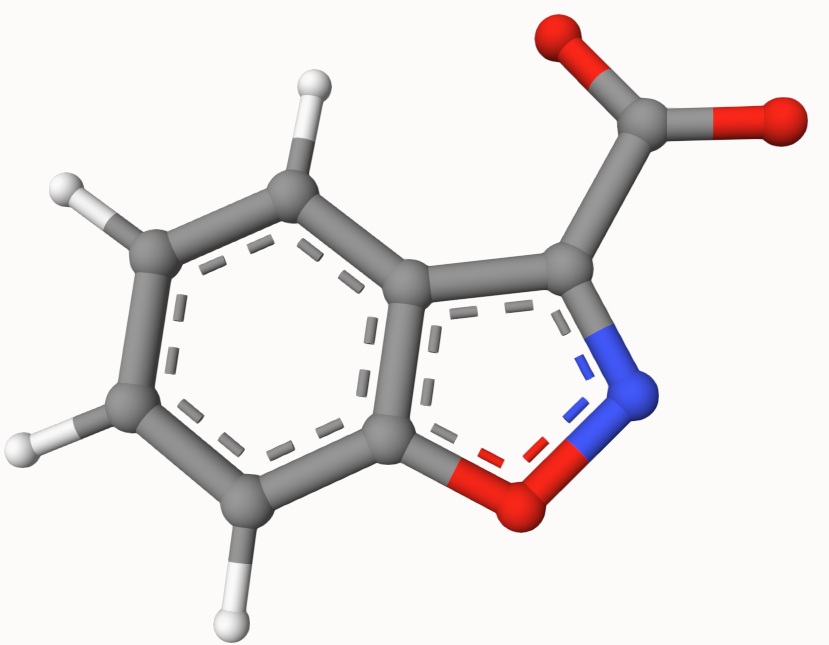
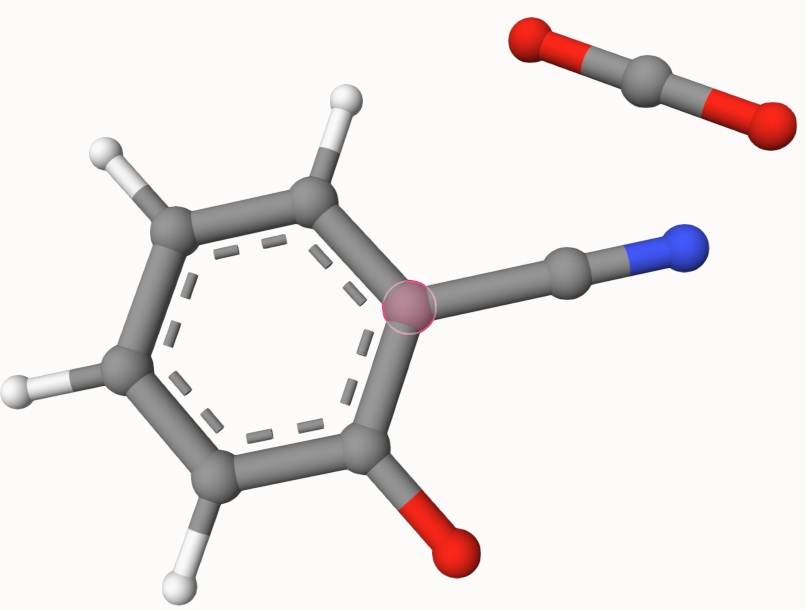
The optimized transition state (ts-2-opt.xyz) and the reaction path (ts-2-opt-traj.xyz) are shown below:
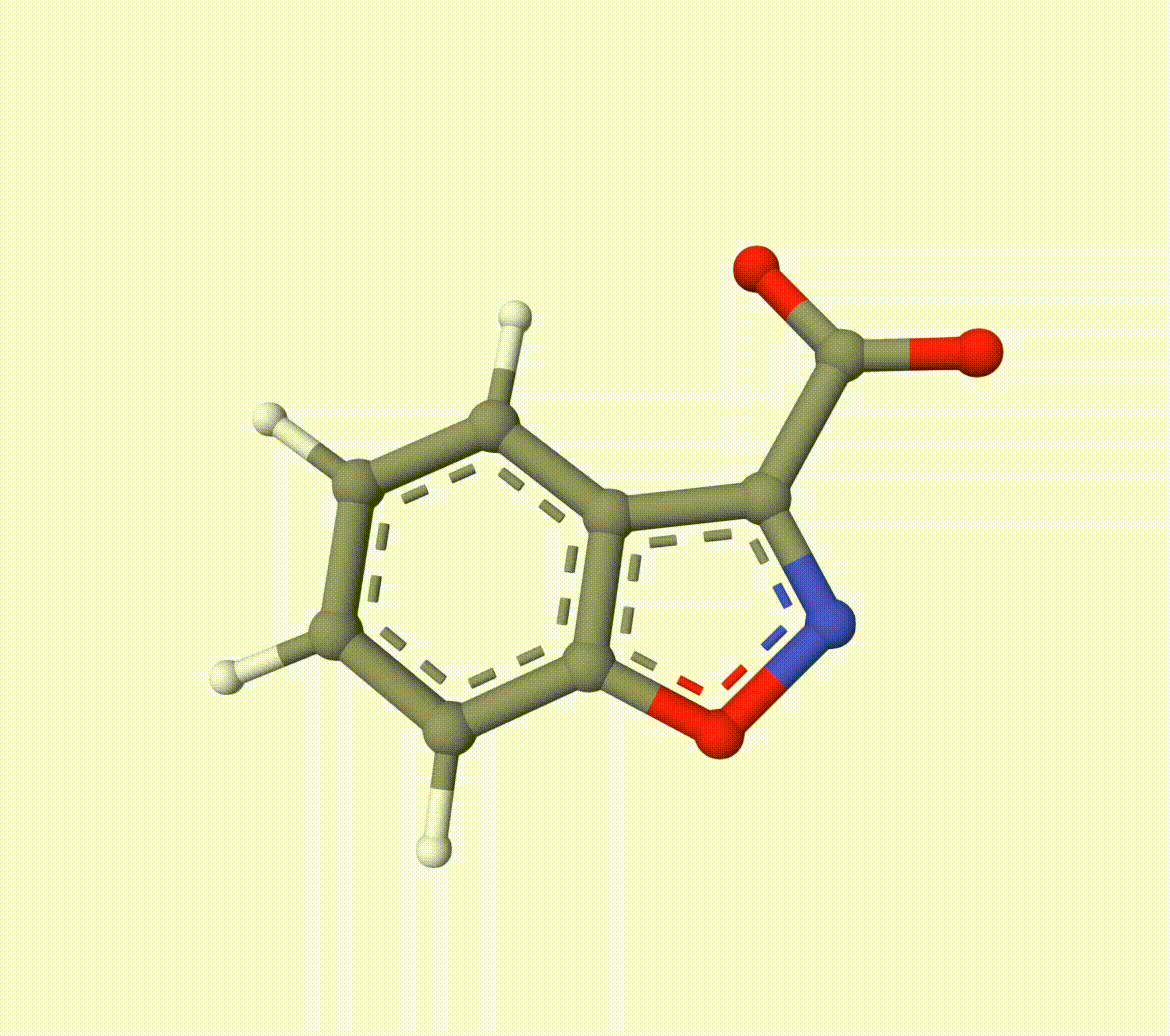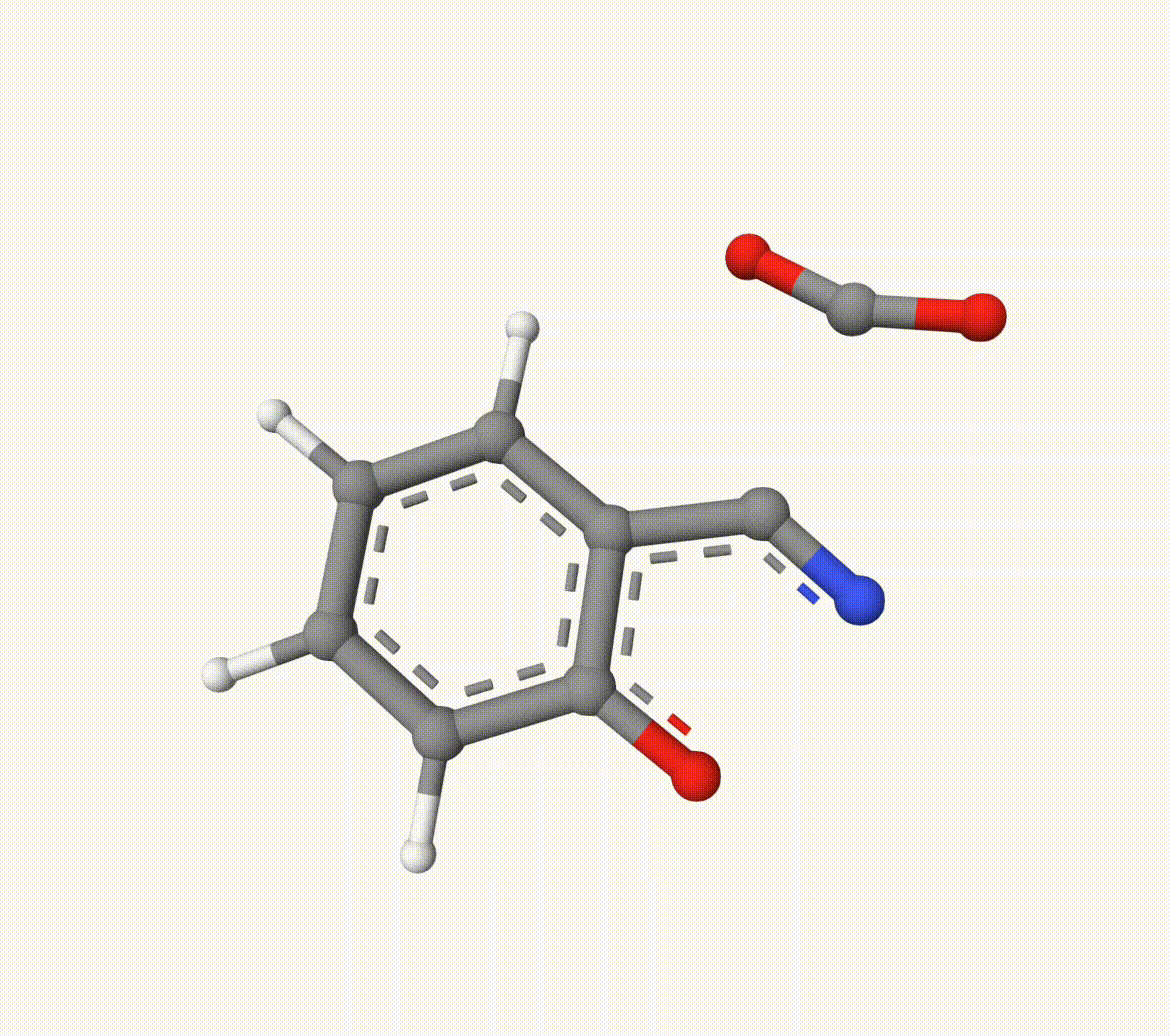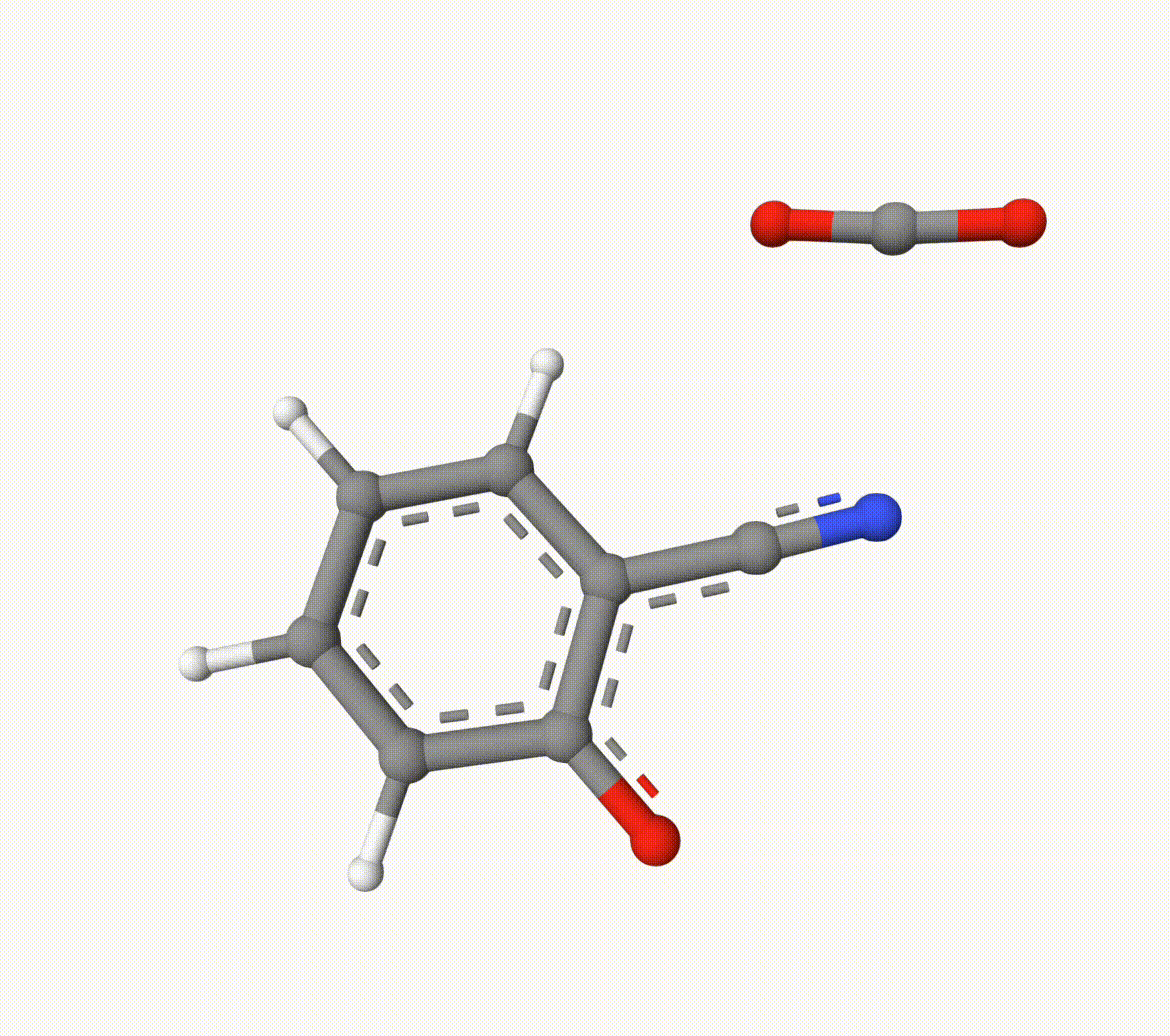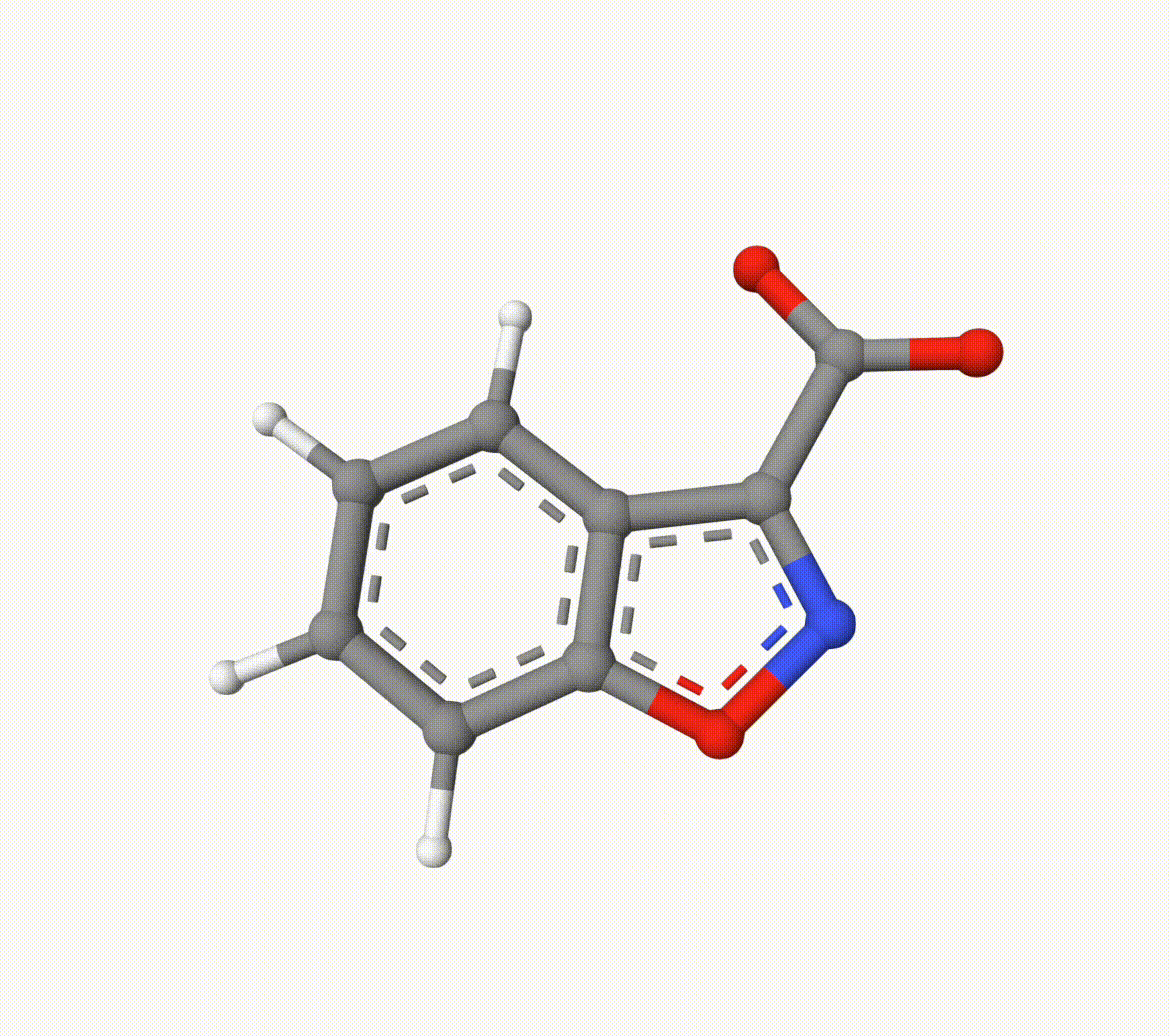
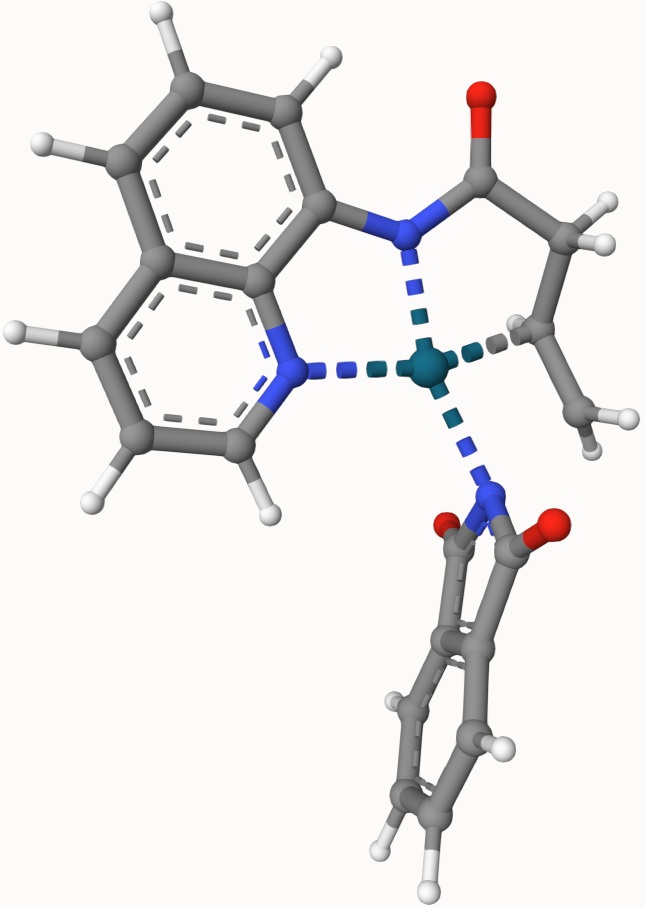
Example: Transition State of Pd(OAc)-Catalyzed Nucleopalladation
Search for the transition state of the following Pd(OAc)-catalyzed nucleopalladation using the DIMER algorithm at the B3LYP/def2-SVP level of theory:
The structures in a1.xyz and a2.xyz are shown below. They are arranged arbitrarily without prior optimization.
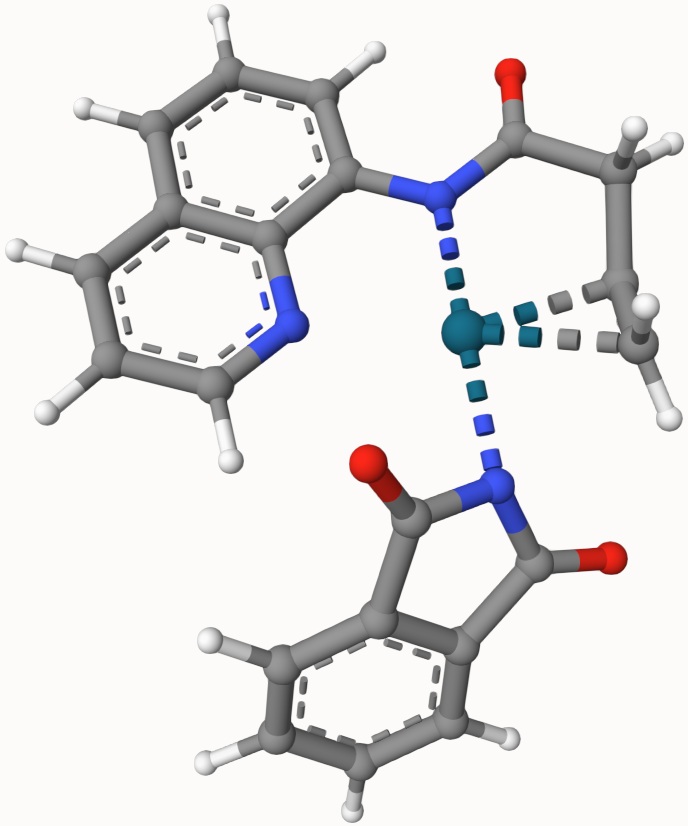
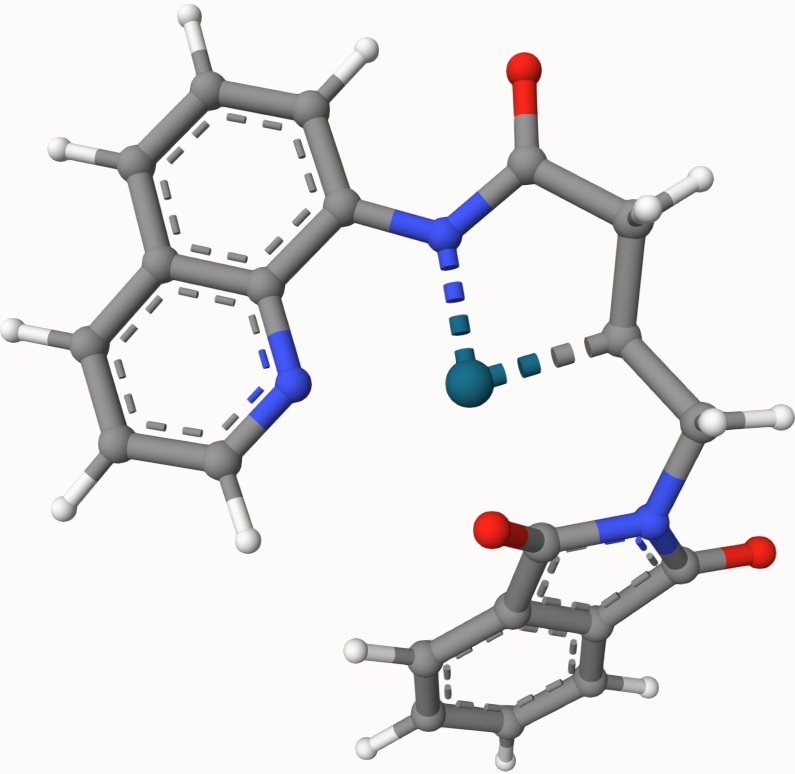
The optimized transition state is provided in ts-3-opt.xyz, as shown below: