All input and output files can be downloaded here.
Molecular Mechanics Energy
This tutorial will lead you step by step to do molecular mechanics (MM) calculations using Qbics. In Qbics, MM means CHARMM potential.
Preface: Topology and Parameters
An MM calculation needs more steps since you must set up topology and parameters for the molecule.
- Topology defines the bonds, angles, dihedrals, impropers and atomic charges. Qbics uses PSF file to record topology.
- Parameters are those for bonding and nonbonding energy terms. Qbics uses CHARMM format parameter files.
Building topology and parameters of a system for MM calculations is probably the most difficult and error-prone step. There are many different ways to build topology for the same system:
- Manually.
- Using ABCluster (http://zhjun-sci.com/software-abcluster-download.php).
- Using VMD (http://www.ks.uiuc.edu/Research/vmd/).
- Using CHARMM-GUI (https://charmm-gui.org/).
In this tutorial, we will use CHARMM-GUI and VMD, but you can use your familiar tools to do this job.
Organic Molecules
Let's consider abscisic acid (ABA), the structure being
aba.xyz
38
ABA
C 23.311 -18.840 -14.802
O 22.378 -18.623 -15.594
C 27.124 -18.220 -14.346
H 27.261 -17.627 -15.282
H 27.794 -19.116 -14.381
H 27.431 -17.592 -13.471
C 26.435 -23.603 -14.235
H 25.321 -23.539 -14.315
H 26.732 -24.676 -14.326
H 26.877 -23.044 -15.098
C 23.639 -20.220 -11.312
H 23.690 -21.300 -11.591
H 24.389 -20.029 -10.506
H 22.617 -19.992 -10.925
C 23.717 -17.911 -12.038
H 22.674 -17.796 -11.656
H 24.434 -17.656 -11.220
H 23.872 -17.199 -12.884
C 22.971 -19.686 -13.625
H 21.885 -19.560 -13.374
H 23.130 -20.759 -13.921
C 23.951 -19.349 -12.504
C 26.897 -23.025 -12.961
C 25.697 -18.663 -14.167
C 24.698 -18.331 -15.020
H 24.854 -17.761 -15.949
C 25.719 -20.928 -13.379
H 25.261 -21.328 -14.295
C 26.520 -21.655 -12.623
H 26.783 -21.358 -11.583
C 27.742 -23.786 -12.232
H 27.976 -24.813 -12.560
C 28.243 -23.366 -10.915
O 28.682 -24.260 -10.152
O 28.189 -22.178 -10.494
C 25.393 -19.506 -12.949
O 26.219 -18.945 -11.886
H 27.080 -19.387 -11.914 For a general organic molecule, we recommend to use CHARMM-GUI to build topology.
- Save it to MOL format using, say, GaussView.
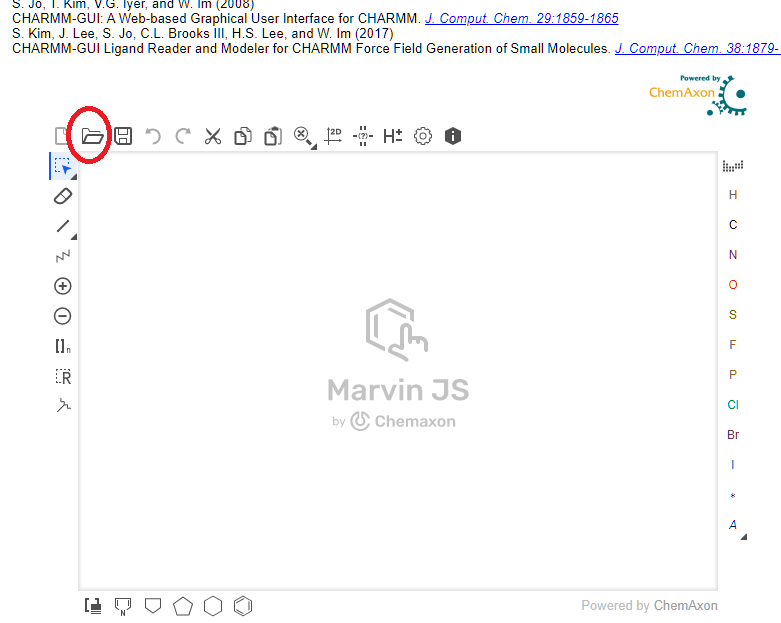
- Open https://charmm-gui.org/, click
Input Generator, thenLigand Reader & Modeler. Open this MOL file in the building part.
- In the edit box, the red ones are chemically incorrect. Fix them before proceeding with your chemical knowledge.
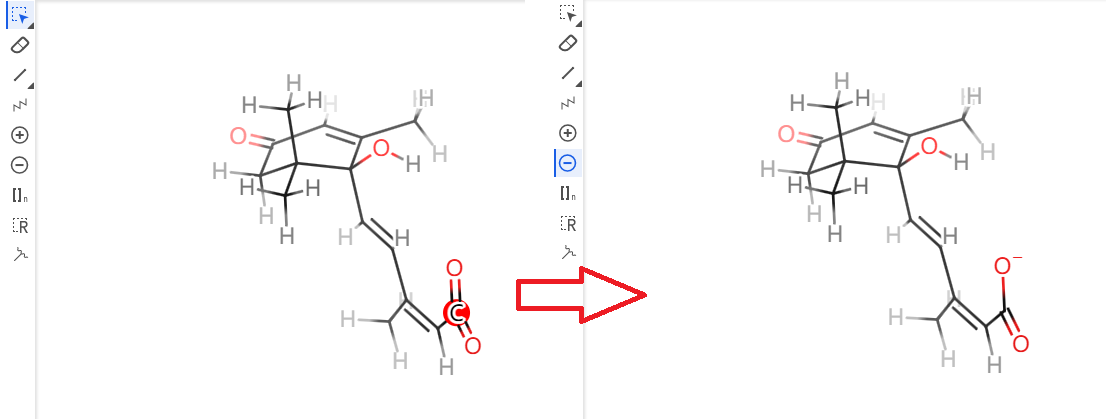
- Click
Next Step. Let's name this ligandABA. After several steps, you will be able to download the generated files by clickingdownload .tgz.
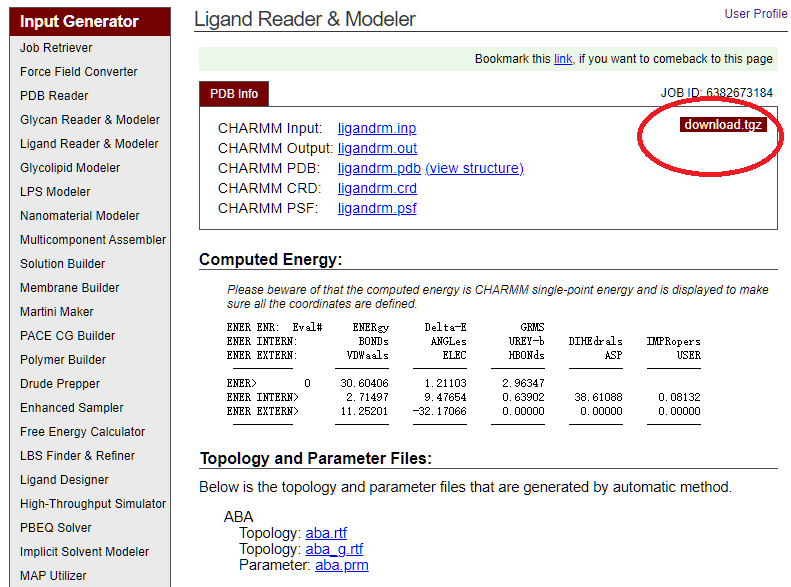
In the compressed package, there are files that are suitable for AMBER, GROMACS, NAMD, CHAMMR, and of course, Qbics. Some useful ones are:
ligandrm.psfThe topology of the molecule that can be read by Qbics.aba/aba.rtfThe topology of the molecule that can be used in VMD.ligandrm.pdbThe coordinates of the molecule in PDB file.aba/aba.prmThe CHARMM parameter files for this molecule. If you open it, you can see the following lines:
aba/aba.prm
...
G2DC1 CG2DC1 CG2O5 CG321 1.4000 2 180.00 ! aba , from CG2DC3 CG2DC1 CG2O5 CG331, penalty= 4.4
HGA4 CG2DC1 CG2O5 CG321 0.0000 2 180.00 ! aba , from HGA4 CG2DC1 CG2O5 CG331, penalty= 0.9
CG2DC1 CG2DC1 CG301 CG2DC2 0.0000 3 180.00 ! aba , from CG2D1O CG2DC1 CG321 CG2D1, penalty= 15.5
...These lines are the CHARMM force field parameters guessed by CHARMM-GUI. Note that, the parameters with larger penalty have a lower reliability.
Also, keep in mind that aba/aba.prm ONLY has the parameters for this molecule. We also need a general CHARMM force field parameter file. For general organic molecules, the CGenFF is a suitable choice. This force field is available here:
toppar/par_all36_cgenff.prm
These files will be used in the following examples.
Biomolecules
For biomolecules, the generation of force field parameters and topology has become a routine job, so it is (probably) easier than organic molecules.
You are encouraged to learn to contruct biomolecular topology with both CHARMM-GUI and VMD.
We will take a protein "ubiquitin" (PDB ID: 1UBQ) and use VMD as an example.
- Open https://charmm-gui.org/, click
Input Generator, thenSolution Builder. Type1UBQinDownload PDB File:. ClickNext Step. - Select only
Protein(discard water). ClickNext Step. - Click
Next Step. - Now you can download protein topology and parameters by clicking
download .tgz. But we want to calculate a protein in solution. So, you can adjust protein solution options on this page. In this case, nothing needs to be changed. ClickNext Step. - Click
Next Stepto download the system. We get a system of protein, water, and some ions to neutralize the solution and provide ionic strength. Note that, the page shows that the 3 lengths of this solution box is64,64, and64.
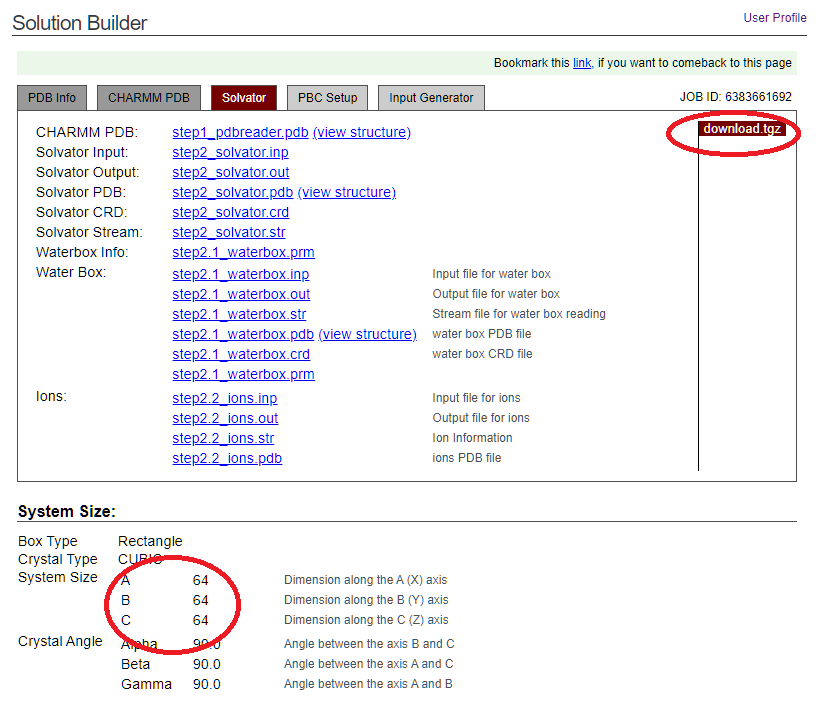
In the compressed package, the files that are useful for us are:
step2_solvator.pdbThe structure of the solvated protein.step2_solvator.psfThe topology of the solvated protein.toppar/par_all36m_prot.prmThe parameters of proteins.toppar/toppar_water_ions.strThe parameters of ions.
The two systems generated above are shown here:
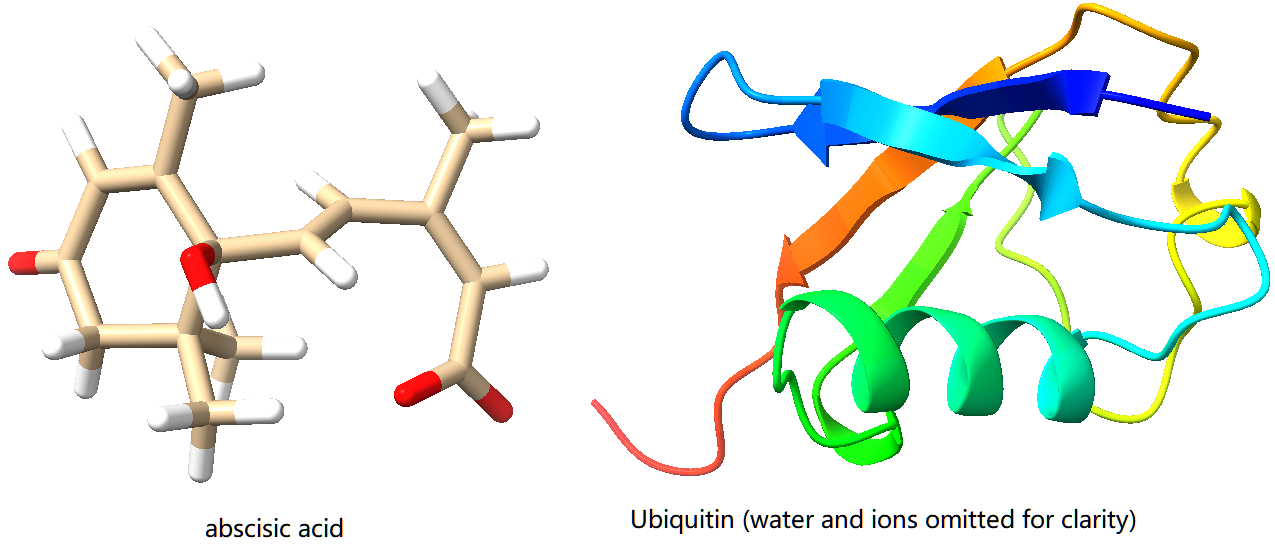
Other Molecules
For other molecules, like inorganic materials, building their topology or fitting force field parameters are rather nontrivial and case-dependend.
Periodic MM Calculations
We will do an MM calculation for the ubituitin in water now.
- Copy
step2_solvator.pdb,step2_solvator.psf,toppar/par_all36m_prot.prm, andtoppar/toppar_water_ions.strto the current path.
For CHARMM files, par* usually contains only parameters, top* usually contains only topologies, but toppar* contains both parameters and topologies. Qbics often fails to parse toppar*, so you have to delete topology information in toppar* and keep only parameter information.
- Open
toppar_water_ions.str, delete lines 1-187 so topology information is removed and only parameter information is reserved. - Prepare the following input file:
protein.inp
charmm
parameters par_all36m_prot.prm toppar_water_ions.str
topology step
scaling14 1.0
rcutoff 12.0
rswitch 10.0
use_PBC
Lbox 64 64 64
electrostatic pme
PMEk 64 64 64
end
mol
step2_solvator.pdb
end
task
energy charmm
endLet's explain this file line-by-line.
- In
mol ... end, you can input a PDB file name, or XYZ file name, or just write coordinates explicitly. task ... endThis tells Qbics what it should do.energy charmmmeans it will use CHARMM force field to calculate energy. You can put several tasks in this block.- MM calculations need only
charmm ... endblock.
parametersThe CHARMM parameter files. You can provide many ones.topologyThe topology of the system in PSF format.scaling14The scaling factor for 1-4 interactions. Usually1.0or0.5is used.rcutoffandrswitchThe cutoff and switch distance (in Angstrom) for nonbonded interactions.
- For van der Waals interactions, they are the switch and cutoff distances for the switching function.
- For electrostatic interaction energies: when
electrostaticiscutoff, they are the switch and cuttoff distances for its switching function; whenelectrostaticispme,rcutoffis the cutoff distance for short-range part, andrswitchis not used.
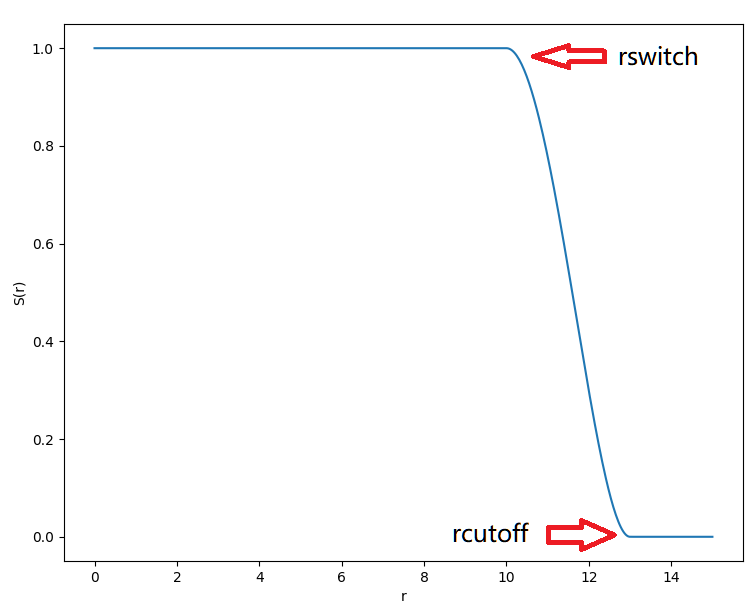
use_pbcThe periodic boundary condition (PBC) is used. If it is not present, a gas phase calculation is applied.LboxThe 3 lengths (in Angstrom) of the simulation box. Only valid whenuse_pbcis applied.electrostaticThe calculation method of electrostatic energy, which can be cut off at a certain distance, or particle mesh Ewald (PME) algorithm. For gas phase calculations, onlycutoffis allowed; when PBC is turned on, one can use eitherpme(you should almost ALWAY use this) orcutoff(NOT recommended).PMEkThe number of K points on 3 lengths of the box for PME calculations. It is recommended to be an integer that is close to the box length and is factorized to only 2, 3, and 5.
For example, the box lengths are 34, 47, and 51, then one can set PMEk to:
- 36 = 2 × 2 × 3 × 3
- 48 = 2 × 2 × 2 × 2 × 3
- 54 = 2 × 3 × 3 × 3
You should NOT use 51, since it is factorized to 3x17.
Now you can run the calculation:
$ Qbics protein.inp -n 4 > protein.outIn protein.out, you can find the calculated energies:
protein.out
CHARMM energies
===============
Bond energy: 193.41739340 kcal/mol
Angle energy: 297.22428926 kcal/mol
Dihedral energy: 730.60163878 kcal/mol
Improper energy: 10.70613120 kcal/mol
Coulombic energy: -78515.73841968 kcal/mol
Lennard-Jones energy: 376696.95829744 kcal/mol
---------------------------------------------------
CHARMM energy: 299413.16933040 kcal/mol
Final total energy: 299413.16933040 kcal/molGas Phase MM Calculations
We will do an MM calculation for abscisic acid in gas phase now.
- Copy
ligandrm.pdb,ligandrm.psf,toppar/aba.prm, andtoppar/par_all36_cgenff.prmto the current path. - Prepare the following input file:
aba.inp
charmm
parameters par_all36_cgenff.prm aba.prm
topology ligandrm.psf
scaling14 1.0
rcutoff 12.0
rswitch 13.0
end
mol
ligandrm.pdb
end
task
energy charmm
end- In this input file, note that we do not write
use_pbc, indicating that no PBC is applied, so this is a gas phase calculation. We do not writeelectrostaticsince the default option for gas phase calculation iscutoff. - You may note that
rswitchis larger thanrcutoff. In Qbics, this means that no cutoff is applied: all pair-wise interactions will be calculated. This is recommended in gas phase MM calculations.
You can run the calculation by this:
$ Qbics aba.inp -n 4 > aba.outThe end of aba.out is:
aba.out
CHARMM energies
===============
Bond energy: 2.74396036 kcal/mol
Angle energy: 10.10698184 kcal/mol
Dihedral energy: 35.62171511 kcal/mol
Improper energy: 0.08140330 kcal/mol
Coulombic energy: -49.04404522 kcal/mol
Lennard-Jones energy: 11.20494529 kcal/mol
---------------------------------------------------
CHARMM energy: 10.71496067 kcal/mol