Tip
All input files can be downloaded: Files.
tddft
This keyword defines how to perform time-dependent DFT (TDDFT) calculation.
Options
- type
Value
TDDFTStandard TDDFT.TDATDDFT with Tamm-Dancoff approximation (TDA).RTDDFTRestricted TDDFT.UTDDFTUnrestricted TDDFT.XTDDFTSpin-adaptive TDDFT.RTDARestricted TDA.UTDAUnrestricted TDA.XTDASpin-adaptive TDA.Default
TDDFTThe type of TDDFT calculations. TDA is an approximate case of TDDFT. For closed-shell systems,
RTDDFTandRTDAare the best choices. For closed- and open-shell systems,UTDDFTandUTDAare available. For open-shell systems,XTDDFTandXTDAare also available. IfTDDFTorTDAis used, a suitable method will be automatically selected.
- spin_flip
Value
NoneNo spin flipping is performed.UpFlip beta electrons to alpha ones.DownFlip alpha electrons to beta ones.Default
NoneSpin flipping way for the excited states.
- num_states
Value
An integer.
Default
5The number of excited states to be calculated.
- max_it
Value
An integer.
Default
100The maximum number of Davidson iterations for the TDDFT calculation. If TDDFT fails to converge, you can try to increase this number.
- dim_trials
Value
An integer.
Default
50The maximum dimension of trial vectors. If TDDFT fails to converge, you can try to increase this number.
- energy_cov
Value
A real number.
Default
1.E-7The energy convergence threshold for the TDDFT calculation.
- vec_cov
Value
A real number.
Default
1.E-5The excited state vector convergence threshold for the TDDFT calculation.
- precondition_threshold
Value
A real number.
Default
1.E-8The precondition convergence threshold for the TDDFT calculation.
- print_coeff_threshold
Value
A real number.
Default
0.01Print the excited state coefficients when the absolute value is larger than this threshold.
- transxtdvec
Transform the excited state vectors from alpha/beta to spin-adapted ones.
Theoretical Background
Time-dependent DFT (TDDFT) is a powerful tool for studying the electronic structure of molecules and materials. It is a time-dependent extension of density functional theory (DFT), which allows for the calculation of excited states and transition properties.
Input Examples
Example: TDDFT Calculation of HCHO
In this example, we will perform a TDDFT calculation of HCHO at TD-B3LYP/cc-pVTZ level of theory. The input file is as follows:
1basis
2 cc-pvtz
3end
4
5scf
6 charge 0
7 spin2p1 1
8 type R
9end
10
11mol
12 C -0.000756 -0.520733 0.
13 H 0.935697 -1.111766 0.
14 H -0.939631 -1.107897 0.
15 O 0.001792 0.678123 0
16end
17
18tddft
19 type tddft
20 num_states 10
21 max_it 100
22 dim_trials 50
23 print_coeff_threshold 0.01
24end
25
26task
27 tddft b3lyp
28end
After running the calculation, you will find the following lines:
1#1: Absolute energy = -114.39668857 Hartree
2 Excited energy = 4.1600 eV; wavelength = 298.04 nm, oscillator strength = 0.0000
3 Transition dipole moment (a.u.): -0.00000 -0.00000 -0.00000
4 CV(0) 8 --> 9: Coefficient = -0.9512, Percentage = 99.7 %, IPA = 6.0538 eV
5#2: Absolute energy = -114.26455102 Hartree
6 Excited energy = 7.7556 eV; wavelength = 159.86 nm, oscillator strength = 0.0917
7 Transition dipole moment (a.u.): 0.69575 -0.00144 -0.00000
8 CV(0) 8 --> 10: Coefficient = 0.9803, Percentage = 99.1 %, IPA = 8.7346 eV
9#3: Absolute energy = -114.20726662 Hartree
10 Excited energy = 9.3144 eV; wavelength = 133.11 nm, oscillator strength = 0.0004
11 Transition dipole moment (a.u.): 0.00000 -0.00000 0.03936
12 CV(0) 6 --> 9: Coefficient = 0.9615, Percentage = 99.0 %, IPA = 11.0312 eV
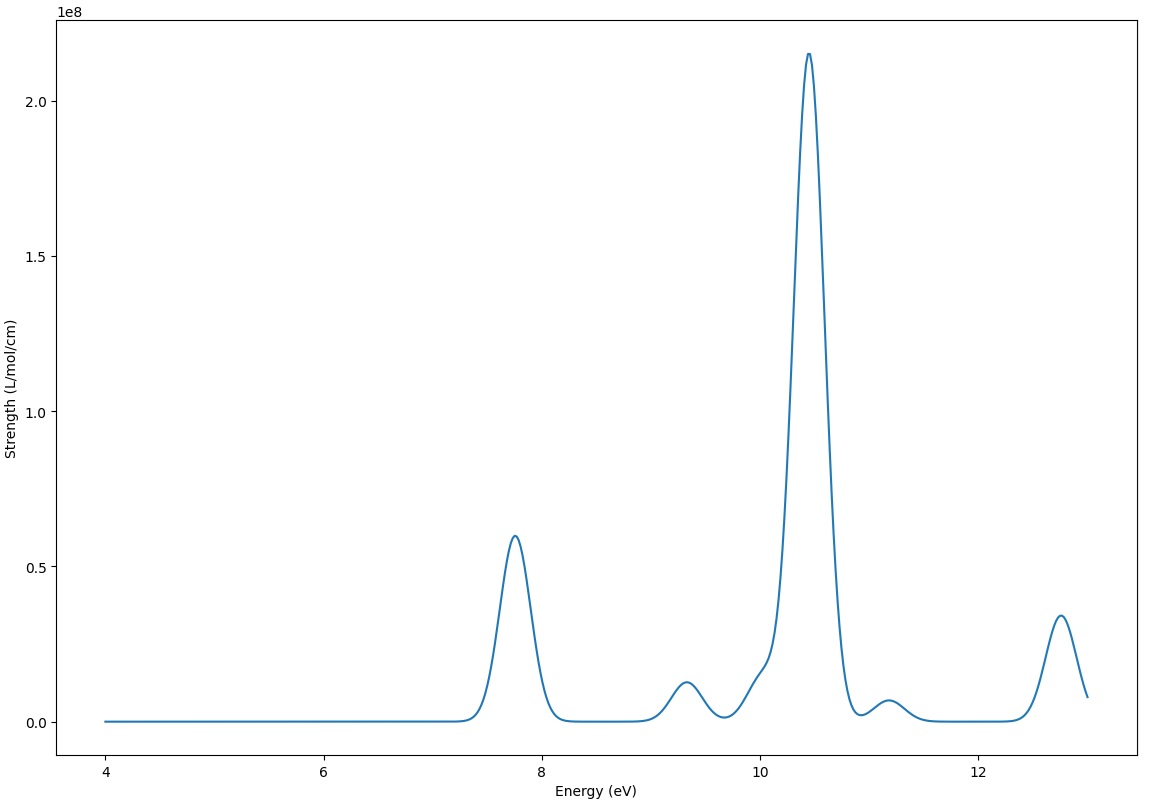
You can find excited state energies, oscillator strengths, transition dipole moments, and orbital transition coefficients.
You can also find the spectrum file tddft-1-spectrum.txt.
You can plot the spectrum using a script provided by Qbics, i.e., tools/plotspec.py. Copy this file to the same directory as the input file, and modify the following parameters:
1if __name__ == "__main__":
2 fn = "tddft-1-spectrum.txt" # Spectrum file name.
3 eL_eV = 4 # Lower energy limit.
4 eH_eV = 13 # Higher energy limit.
5 sigma_eV = 0.2 # Sigma value.
6 num_ps = 500 # Number of points.
7 use_angle = False # Whether to use angle dependence.
8 incident_angles = [i*30 for i in range(4)] # Incident angles.
Line 2: Change the spectrum file name.
Line 3,4: Change the lower and upper energy limit.
Line 5: Change the sigma value.
Line 6: Change the number of points. The larger the number, the smoother the spectrum.
Line 7: Whether to use angle dependence.
Line 8: Incident angles.
Then, you can run the script using the following command:
1$ python plotspec.py
You will find following spectrum plot: